Ghoncheh Mashayekhi , John Vant , Abhigna Polavarapu , Abbas Ourmazd , Abhishek Singharoy
{"title":"Energy landscape of the SARS-CoV-2 reveals extensive conformational heterogeneity","authors":"Ghoncheh Mashayekhi , John Vant , Abhigna Polavarapu , Abbas Ourmazd , Abhishek Singharoy","doi":"10.1016/j.crstbi.2022.02.001","DOIUrl":null,"url":null,"abstract":"<div><p>Cryo-electron microscopy (cryo-EM) has produced a number of structural models of the SARS-CoV-2 spike, already prompting biomedical outcomes. However, these reported models and their associated electrostatic potential maps represent an unknown admixture of conformations stemming from the underlying energy landscape of the spike protein. As with any protein, some of the spike's conformational motions are expected to be biophysically relevant, but cannot be interpreted only by static models. Using experimental cryo-EM images, we present the energy landscape of the glycosylated spike protein, and identify the diversity of low-energy conformations in the vicinity of its open (so called 1RBD-up) state. The resulting atomic refinement reveal global and local molecular rearrangements that cannot be inferred from an average 1RBD-up cryo-EM model. Here we report varied degrees of “openness” in global conformations of the 1RBD-up state, not revealed in the single-model interpretations of the density maps, together with conformations that overlap with the reported models. We discover how the glycan shield contributes to the stability of these low-energy conformations. Five out of six binding sites we analyzed, including those for engaging ACE2, therapeutic mini-proteins, linoleic acid, two different kinds of antibodies, switch conformations between their known apo- and holo-conformations, even when the global spike conformation is 1RBD-up. This apo-to-holo switching is reminiscent of a conformational preequilibrium. We found only one binding site, namely that of AB-C135 remains in apo state within all the sampled free energy-minimizing models, suggesting an induced fit mechanism for the docking of this antibody to the spike.</p></div>","PeriodicalId":10870,"journal":{"name":"Current Research in Structural Biology","volume":"4 ","pages":"Pages 68-77"},"PeriodicalIF":2.7000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/4f/91/main.PMC8902891.pdf","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Research in Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2665928X22000034","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 3
Abstract
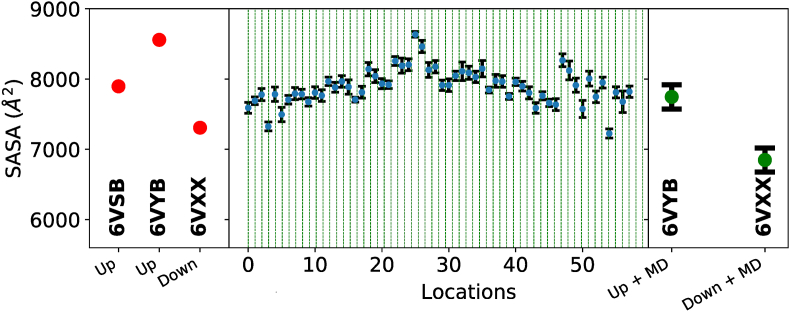
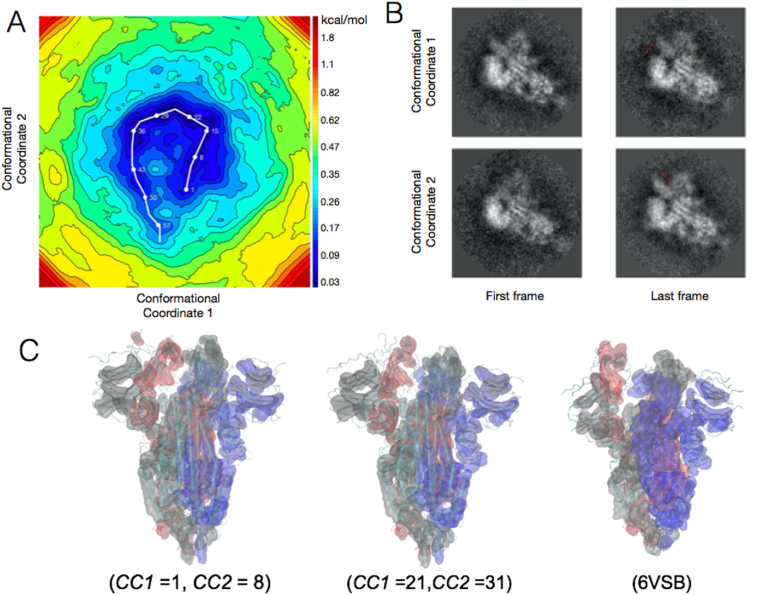
Cryo-electron microscopy (cryo-EM) has produced a number of structural models of the SARS-CoV-2 spike, already prompting biomedical outcomes. However, these reported models and their associated electrostatic potential maps represent an unknown admixture of conformations stemming from the underlying energy landscape of the spike protein. As with any protein, some of the spike's conformational motions are expected to be biophysically relevant, but cannot be interpreted only by static models. Using experimental cryo-EM images, we present the energy landscape of the glycosylated spike protein, and identify the diversity of low-energy conformations in the vicinity of its open (so called 1RBD-up) state. The resulting atomic refinement reveal global and local molecular rearrangements that cannot be inferred from an average 1RBD-up cryo-EM model. Here we report varied degrees of “openness” in global conformations of the 1RBD-up state, not revealed in the single-model interpretations of the density maps, together with conformations that overlap with the reported models. We discover how the glycan shield contributes to the stability of these low-energy conformations. Five out of six binding sites we analyzed, including those for engaging ACE2, therapeutic mini-proteins, linoleic acid, two different kinds of antibodies, switch conformations between their known apo- and holo-conformations, even when the global spike conformation is 1RBD-up. This apo-to-holo switching is reminiscent of a conformational preequilibrium. We found only one binding site, namely that of AB-C135 remains in apo state within all the sampled free energy-minimizing models, suggesting an induced fit mechanism for the docking of this antibody to the spike.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


