{"title":"An objective criterion to evaluate sequence-similarity networks helps in dividing the protein family sequence space.","authors":"Bastian Volker Helmut Hornung, Nicolas Terrapon","doi":"10.1371/journal.pcbi.1010881","DOIUrl":null,"url":null,"abstract":"<p><p>The deluge of genomic data raises various challenges for computational protein annotation. The definition of superfamilies, based on conserved folds, or of families, showing more recent homology signatures, allow a first categorization of the sequence space. However, for precise functional annotation or the identification of the unexplored parts within a family, a division into subfamilies is essential. As curators of an expert database, the Carbohydrate Active Enzymes database (CAZy), we began, more than 15 years ago, to manually define subfamilies based on phylogeny reconstruction. However, facing the increasing amount of sequence and functional data, we required more scalable and reproducible methods. The recently popularized sequence similarity networks (SSNs), allows to cope with very large families and computation of many subfamily schemes. Still, the choice of the optimal SSN subfamily scheme only relies on expert knowledge so far, without any data-driven guidance from within the network. In this study, we therefore decided to investigate several network properties to determine a criterion which can be used by curators to evaluate the quality of subfamily assignments. The performance of the closeness centrality criterion, a network property to indicate the connectedness within the network, shows high similarity to the decisions of expert curators from eight distinct protein families. Closeness centrality also suggests that in some cases multiple levels of subfamilies could be possible, depending on the granularity of the research question, while it indicates when no subfamily emerged in some family evolution. We finally used closeness centrality to create subfamilies in four families of the CAZy database, providing a finer functional annotation and highlighting subfamilies without biochemically characterized members for potential future discoveries.</p>","PeriodicalId":49688,"journal":{"name":"PLoS Computational Biology","volume":null,"pages":null},"PeriodicalIF":4.3000,"publicationDate":"2023-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10461819/pdf/","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"PLoS Computational Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1371/journal.pcbi.1010881","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 2
Abstract
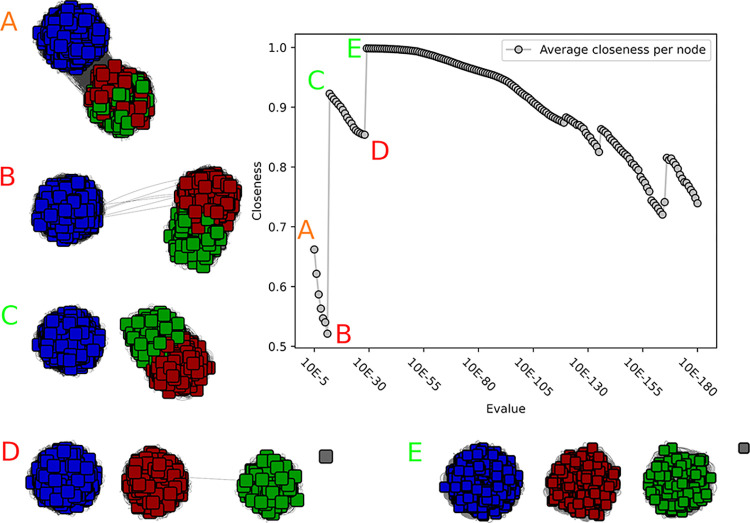
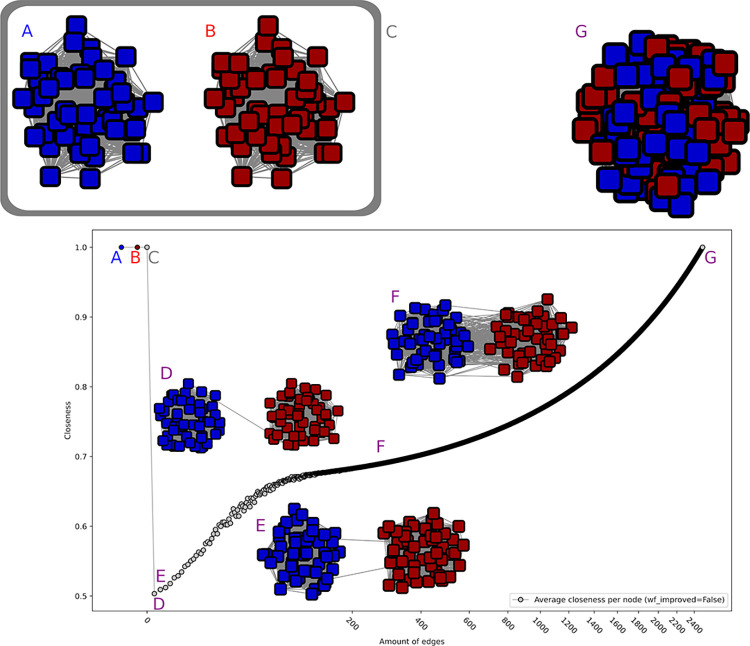
The deluge of genomic data raises various challenges for computational protein annotation. The definition of superfamilies, based on conserved folds, or of families, showing more recent homology signatures, allow a first categorization of the sequence space. However, for precise functional annotation or the identification of the unexplored parts within a family, a division into subfamilies is essential. As curators of an expert database, the Carbohydrate Active Enzymes database (CAZy), we began, more than 15 years ago, to manually define subfamilies based on phylogeny reconstruction. However, facing the increasing amount of sequence and functional data, we required more scalable and reproducible methods. The recently popularized sequence similarity networks (SSNs), allows to cope with very large families and computation of many subfamily schemes. Still, the choice of the optimal SSN subfamily scheme only relies on expert knowledge so far, without any data-driven guidance from within the network. In this study, we therefore decided to investigate several network properties to determine a criterion which can be used by curators to evaluate the quality of subfamily assignments. The performance of the closeness centrality criterion, a network property to indicate the connectedness within the network, shows high similarity to the decisions of expert curators from eight distinct protein families. Closeness centrality also suggests that in some cases multiple levels of subfamilies could be possible, depending on the granularity of the research question, while it indicates when no subfamily emerged in some family evolution. We finally used closeness centrality to create subfamilies in four families of the CAZy database, providing a finer functional annotation and highlighting subfamilies without biochemically characterized members for potential future discoveries.
期刊介绍:
PLOS Computational Biology features works of exceptional significance that further our understanding of living systems at all scales—from molecules and cells, to patient populations and ecosystems—through the application of computational methods. Readers include life and computational scientists, who can take the important findings presented here to the next level of discovery.
Research articles must be declared as belonging to a relevant section. More information about the sections can be found in the submission guidelines.
Research articles should model aspects of biological systems, demonstrate both methodological and scientific novelty, and provide profound new biological insights.
Generally, reliability and significance of biological discovery through computation should be validated and enriched by experimental studies. Inclusion of experimental validation is not required for publication, but should be referenced where possible. Inclusion of experimental validation of a modest biological discovery through computation does not render a manuscript suitable for PLOS Computational Biology.
Research articles specifically designated as Methods papers should describe outstanding methods of exceptional importance that have been shown, or have the promise to provide new biological insights. The method must already be widely adopted, or have the promise of wide adoption by a broad community of users. Enhancements to existing published methods will only be considered if those enhancements bring exceptional new capabilities.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


