{"title":"Optimal selection of suitable templates in protein interface prediction.","authors":"Steven Grudman, J Eduardo Fajardo, Andras Fiser","doi":"10.1093/bioinformatics/btad510","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Molecular-level classification of protein-protein interfaces can greatly assist in functional characterization and rational drug design. The most accurate protein interface predictions rely on finding homologous proteins with known interfaces since most interfaces are conserved within the same protein family. The accuracy of these template-based prediction approaches depends on the correct choice of suitable templates. Choosing the right templates in the immunoglobulin superfamily (IgSF) is challenging because its members share low sequence identity and display a wide range of alternative binding sites despite structural homology.</p><p><strong>Results: </strong>We present a new approach to predict protein interfaces. First, template-specific, informative evolutionary profiles are established using a mutual information-based approach. Next, based on the similarity of residue level conservation scores derived from the evolutionary profiles, a query protein is hierarchically clustered with all available template proteins in its superfamily with known interface definitions. Once clustered, a subset of the most closely related templates is selected, and an interface prediction is made. These initial interface predictions are subsequently refined by extensive docking. This method was benchmarked on 51 IgSF proteins and can predict nontrivial interfaces of IgSF proteins with an average and median F-score of 0.64 and 0.78, respectively. We also provide a way to assess the confidence of the results. The average and median F-scores increase to 0.8 and 0.81, respectively, if 27% of low confidence cases and 17% of medium confidence cases are removed. Lastly, we provide residue level interface predictions, protein complexes, and confidence measurements for singletons in the IgSF.</p><p><strong>Availability and implementation: </strong>Source code is freely available at: https://gitlab.com/fiserlab.org/interdct_with_refinement.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 9","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10491951/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad510","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: Molecular-level classification of protein-protein interfaces can greatly assist in functional characterization and rational drug design. The most accurate protein interface predictions rely on finding homologous proteins with known interfaces since most interfaces are conserved within the same protein family. The accuracy of these template-based prediction approaches depends on the correct choice of suitable templates. Choosing the right templates in the immunoglobulin superfamily (IgSF) is challenging because its members share low sequence identity and display a wide range of alternative binding sites despite structural homology.
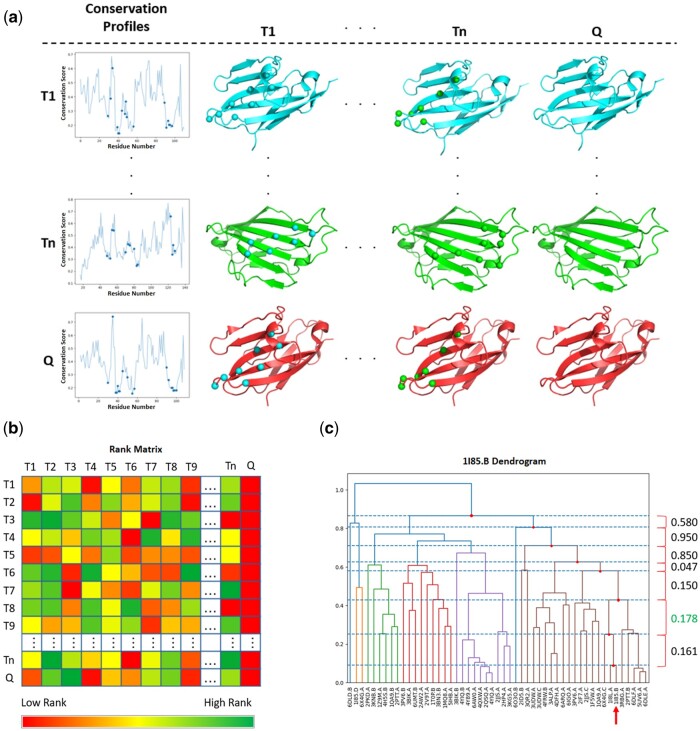
Results: We present a new approach to predict protein interfaces. First, template-specific, informative evolutionary profiles are established using a mutual information-based approach. Next, based on the similarity of residue level conservation scores derived from the evolutionary profiles, a query protein is hierarchically clustered with all available template proteins in its superfamily with known interface definitions. Once clustered, a subset of the most closely related templates is selected, and an interface prediction is made. These initial interface predictions are subsequently refined by extensive docking. This method was benchmarked on 51 IgSF proteins and can predict nontrivial interfaces of IgSF proteins with an average and median F-score of 0.64 and 0.78, respectively. We also provide a way to assess the confidence of the results. The average and median F-scores increase to 0.8 and 0.81, respectively, if 27% of low confidence cases and 17% of medium confidence cases are removed. Lastly, we provide residue level interface predictions, protein complexes, and confidence measurements for singletons in the IgSF.
Availability and implementation: Source code is freely available at: https://gitlab.com/fiserlab.org/interdct_with_refinement.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


