Yunfeng Wang, Haoliang Xue, Marine Aglave, Antoine Lainé, Mélina Gallopin, Daniel Gautheret
{"title":"The contribution of uncharted RNA sequences to tumor identity in lung adenocarcinoma.","authors":"Yunfeng Wang, Haoliang Xue, Marine Aglave, Antoine Lainé, Mélina Gallopin, Daniel Gautheret","doi":"10.1093/narcan/zcac001","DOIUrl":null,"url":null,"abstract":"<p><p>The identity of cancer cells is defined by the interplay between genetic, epigenetic transcriptional and post-transcriptional variation. A lot of this variation is present in RNA-seq data and can be captured at once using reference-free, k-mer analysis. An important issue with k-mer analysis, however, is the difficulty of distinguishing signal from noise. Here, we use two independent lung adenocarcinoma datasets to identify all reproducible events at the k-mer level, in a tumor versus normal setting. We find reproducible events in many different locations (introns, intergenic, repeats) and forms (spliced, polyadenylated, chimeric etc.). We systematically analyze events that are ignored in conventional transcriptomics and assess their value as biomarkers and for tumor classification, survival prediction, neoantigen prediction and correlation with the immune microenvironment. We find that unannotated lincRNAs, novel splice variants, endogenous HERV, Line1 and Alu repeats and bacterial RNAs each contribute to different, important aspects of tumor identity. We argue that differential RNA-seq analysis of tumor/normal sample collections would benefit from this type k-mer analysis to cast a wider net on important cancer-related events. The code is available at https://github.com/Transipedia/dekupl-lung-cancer-inter-cohort.</p>","PeriodicalId":18879,"journal":{"name":"NAR Cancer","volume":"4 1","pages":"zcac001"},"PeriodicalIF":0.0000,"publicationDate":"2022-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/13/fe/zcac001.PMC8807116.pdf","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Cancer","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/narcan/zcac001","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 2
Abstract
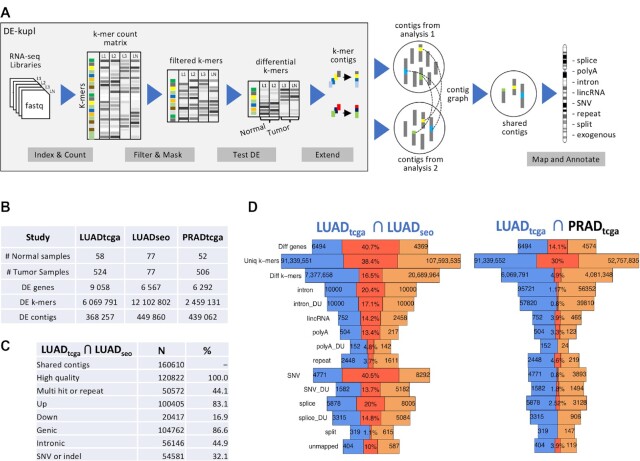
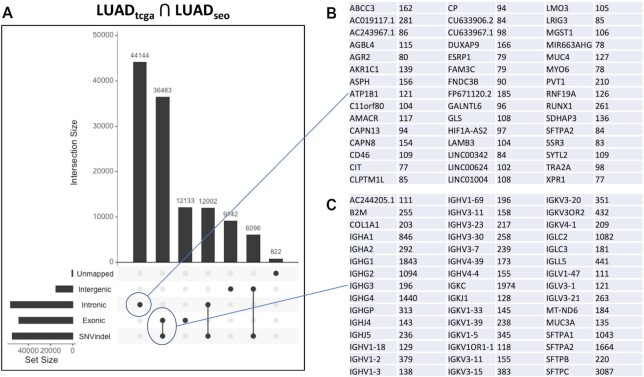
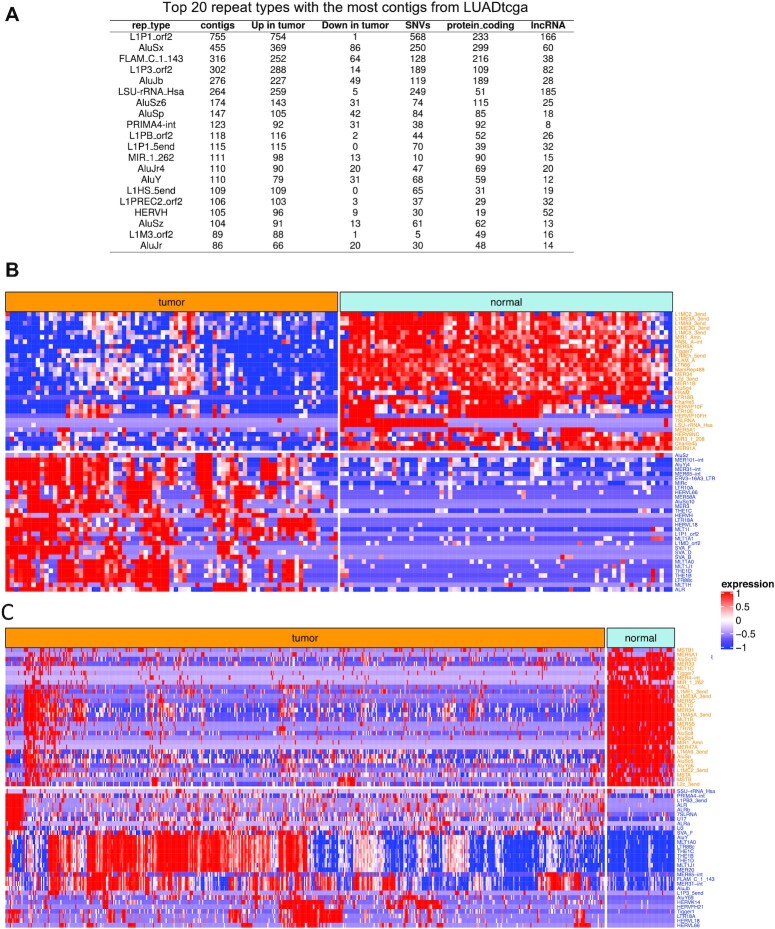
The identity of cancer cells is defined by the interplay between genetic, epigenetic transcriptional and post-transcriptional variation. A lot of this variation is present in RNA-seq data and can be captured at once using reference-free, k-mer analysis. An important issue with k-mer analysis, however, is the difficulty of distinguishing signal from noise. Here, we use two independent lung adenocarcinoma datasets to identify all reproducible events at the k-mer level, in a tumor versus normal setting. We find reproducible events in many different locations (introns, intergenic, repeats) and forms (spliced, polyadenylated, chimeric etc.). We systematically analyze events that are ignored in conventional transcriptomics and assess their value as biomarkers and for tumor classification, survival prediction, neoantigen prediction and correlation with the immune microenvironment. We find that unannotated lincRNAs, novel splice variants, endogenous HERV, Line1 and Alu repeats and bacterial RNAs each contribute to different, important aspects of tumor identity. We argue that differential RNA-seq analysis of tumor/normal sample collections would benefit from this type k-mer analysis to cast a wider net on important cancer-related events. The code is available at https://github.com/Transipedia/dekupl-lung-cancer-inter-cohort.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


