Bryce Kille, Erik Garrison, Todd J Treangen, Adam M Phillippy
{"title":"Minmers are a generalization of minimizers that enable unbiased local Jaccard estimation.","authors":"Bryce Kille, Erik Garrison, Todd J Treangen, Adam M Phillippy","doi":"10.1093/bioinformatics/btad512","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>The Jaccard similarity on k-mer sets has shown to be a convenient proxy for sequence identity. By avoiding expensive base-level alignments and comparing reduced sequence representations, tools such as MashMap can scale to massive numbers of pairwise comparisons while still providing useful similarity estimates. However, due to their reliance on minimizer winnowing, previous versions of MashMap were shown to be biased and inconsistent estimators of Jaccard similarity. This directly impacts downstream tools that rely on the accuracy of these estimates.</p><p><strong>Results: </strong>To address this, we propose the minmer winnowing scheme, which generalizes the minimizer scheme by use of a rolling minhash with multiple sampled k-mers per window. We show both theoretically and empirically that minmers yield an unbiased estimator of local Jaccard similarity, and we implement this scheme in an updated version of MashMap. The minmer-based implementation is over 10 times faster than the minimizer-based version under the default ANI threshold, making it well-suited for large-scale comparative genomics applications.</p><p><strong>Availability and implementation: </strong>MashMap3 is available at https://github.com/marbl/MashMap.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 9","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10505501/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad512","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: The Jaccard similarity on k-mer sets has shown to be a convenient proxy for sequence identity. By avoiding expensive base-level alignments and comparing reduced sequence representations, tools such as MashMap can scale to massive numbers of pairwise comparisons while still providing useful similarity estimates. However, due to their reliance on minimizer winnowing, previous versions of MashMap were shown to be biased and inconsistent estimators of Jaccard similarity. This directly impacts downstream tools that rely on the accuracy of these estimates.
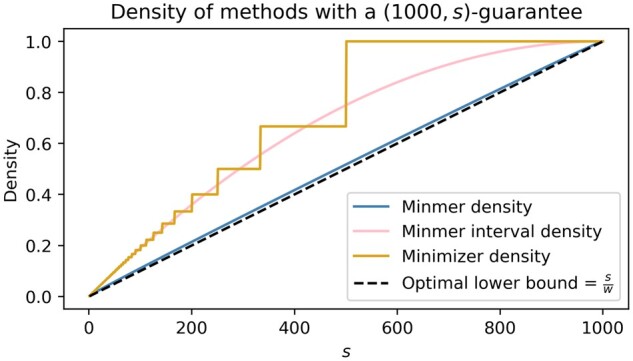
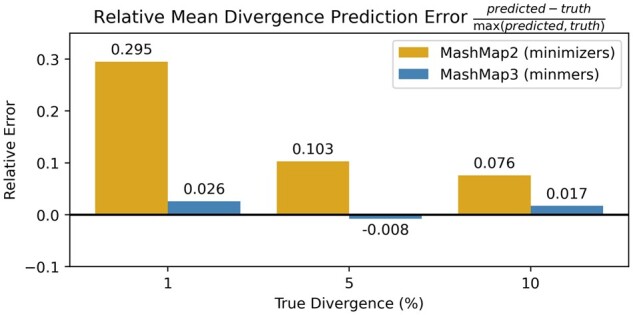
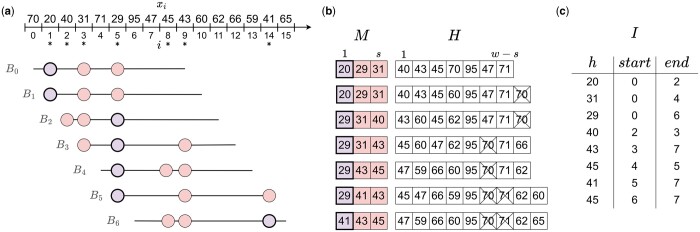
Results: To address this, we propose the minmer winnowing scheme, which generalizes the minimizer scheme by use of a rolling minhash with multiple sampled k-mers per window. We show both theoretically and empirically that minmers yield an unbiased estimator of local Jaccard similarity, and we implement this scheme in an updated version of MashMap. The minmer-based implementation is over 10 times faster than the minimizer-based version under the default ANI threshold, making it well-suited for large-scale comparative genomics applications.
Availability and implementation: MashMap3 is available at https://github.com/marbl/MashMap.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


