E. Fernández-Giménez , M.M. Martínez , R. Marabini , D. Strelak , R. Sánchez-García , J.M. Carazo , C.O.S. Sorzano
{"title":"A new algorithm for particle weighted subtraction to decrease signals from unwanted components in single particle analysis","authors":"E. Fernández-Giménez , M.M. Martínez , R. Marabini , D. Strelak , R. Sánchez-García , J.M. Carazo , C.O.S. Sorzano","doi":"10.1016/j.jsb.2023.108024","DOIUrl":null,"url":null,"abstract":"<div><p>Single particle analysis (SPA) in cryo-electron microscopy (cryo-EM) is highly used to obtain the near-atomic structure of biological macromolecules. The current methods allow users to produce high-resolution maps from many samples. However, there are still challenging cases that require extra processing to obtain high resolution. This is the case when the macromolecule of the sample is composed of different components and we want to focus just on one of them. For example, if the macromolecule is composed of several flexible subunits and we are interested in a specific one, if it is embedded in a viral capsid environment, or if it has additional components to stabilize it, such as nanodiscs. The signal from these components, which in principle we are not interested in, can be removed from the particles using a projection subtraction method. Currently, there are two projection subtraction methods used in practice and both have some limitations. In fact, after evaluating their results, we consider that the problem is still open to new solutions, as they do not fully remove the signal of the components that are not of interest. Our aim is to develop a new and more precise projection subtraction method, improving the performance of state-of-the-art methods. We tested our algorithm with data from public databases and an in–house data set. In this work, we show that the performance of our algorithm improves the results obtained by others, including the localization of small ligands, such as drugs, whose binding location is unknown <em>a priori</em>.</p></div>","PeriodicalId":17074,"journal":{"name":"Journal of structural biology","volume":"215 4","pages":"Article 108024"},"PeriodicalIF":3.0000,"publicationDate":"2023-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of structural biology","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1047847723000874","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
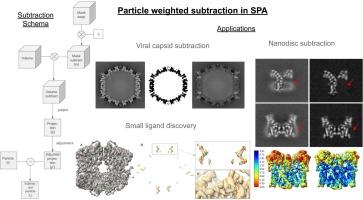
Single particle analysis (SPA) in cryo-electron microscopy (cryo-EM) is highly used to obtain the near-atomic structure of biological macromolecules. The current methods allow users to produce high-resolution maps from many samples. However, there are still challenging cases that require extra processing to obtain high resolution. This is the case when the macromolecule of the sample is composed of different components and we want to focus just on one of them. For example, if the macromolecule is composed of several flexible subunits and we are interested in a specific one, if it is embedded in a viral capsid environment, or if it has additional components to stabilize it, such as nanodiscs. The signal from these components, which in principle we are not interested in, can be removed from the particles using a projection subtraction method. Currently, there are two projection subtraction methods used in practice and both have some limitations. In fact, after evaluating their results, we consider that the problem is still open to new solutions, as they do not fully remove the signal of the components that are not of interest. Our aim is to develop a new and more precise projection subtraction method, improving the performance of state-of-the-art methods. We tested our algorithm with data from public databases and an in–house data set. In this work, we show that the performance of our algorithm improves the results obtained by others, including the localization of small ligands, such as drugs, whose binding location is unknown a priori.
期刊介绍:
Journal of Structural Biology (JSB) has an open access mirror journal, the Journal of Structural Biology: X (JSBX), sharing the same aims and scope, editorial team, submission system and rigorous peer review. Since both journals share the same editorial system, you may submit your manuscript via either journal homepage. You will be prompted during submission (and revision) to choose in which to publish your article. The editors and reviewers are not aware of the choice you made until the article has been published online. JSB and JSBX publish papers dealing with the structural analysis of living material at every level of organization by all methods that lead to an understanding of biological function in terms of molecular and supermolecular structure.
Techniques covered include:
• Light microscopy including confocal microscopy
• All types of electron microscopy
• X-ray diffraction
• Nuclear magnetic resonance
• Scanning force microscopy, scanning probe microscopy, and tunneling microscopy
• Digital image processing
• Computational insights into structure

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


