Mónica C Quiñones-Frías, Dina M Ocken, Avital Rodal
{"title":"Disruption of Synaptic Endoplasmic Reticulum Luminal Protein Containment in <i>Drosophila Atlastin</i> Mutants.","authors":"Mónica C Quiñones-Frías, Dina M Ocken, Avital Rodal","doi":"10.1101/2023.09.01.555994","DOIUrl":null,"url":null,"abstract":"<p><p>The endoplasmic reticulum (ER) extends throughout neurons and regulates many neuronal functions, including neurite outgrowth, neurotransmission, and synaptic plasticity. Mutations in proteins that control ER shape are linked to the neurodegenerative disorder Hereditary Spastic Paraplegia (HSP), yet the ultrastructure and dynamics of neuronal ER remain largely unexplored, especially at presynaptic terminals. Using super-resolution and live imaging in <i>D. melanogaster</i> larval motor neurons, we investigated ER structure at presynaptic terminals of wild-type animals and null mutants of the ER shaping protein and HSP-linked gene, Atlastin. Previous studies using an ER luminal marker reported diffuse localization at <i>Atlastin</i> mutant presynaptic terminals, which was attributed to ER fragmentation. However, using an ER membrane marker, we discovered that <i>Atlastin</i> mutant ER forms robust networks with only mild defects in structural dynamics, indicating the primary defect is functional rather than architectural. We demonstrate that <i>Atlastin</i> mutants progressively displace overexpressed luminal ER proteins to the cytosol during larval development, specifically at synapses, while these proteins remain correctly localized in cell bodies, axons, and muscles. This synaptic-specific displacement phenotype, previously unreported in non-neuronal cells, emphasizes the importance of studying neurons to understand HSP pathogenesis.</p>","PeriodicalId":72407,"journal":{"name":"bioRxiv : the preprint server for biology","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2025-02-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/c6/5c/nihpp-2023.09.01.555994v1.PMC10491308.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"bioRxiv : the preprint server for biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2023.09.01.555994","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
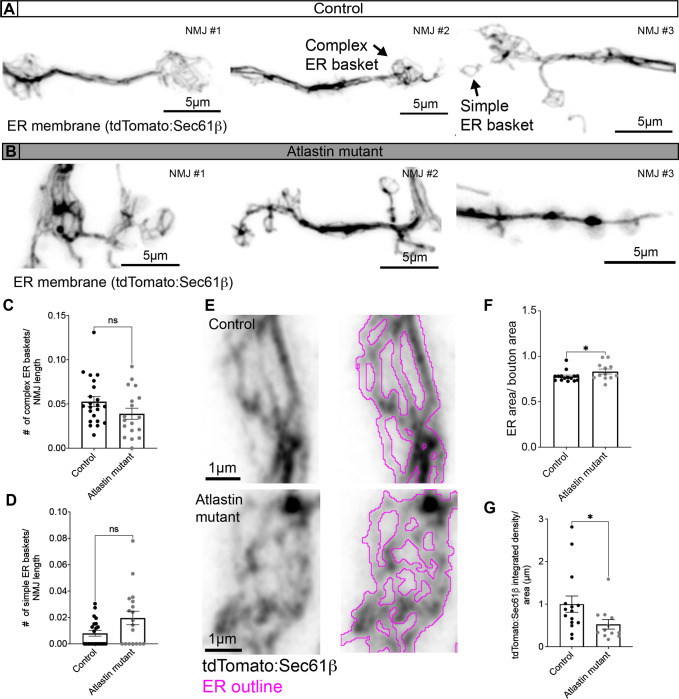
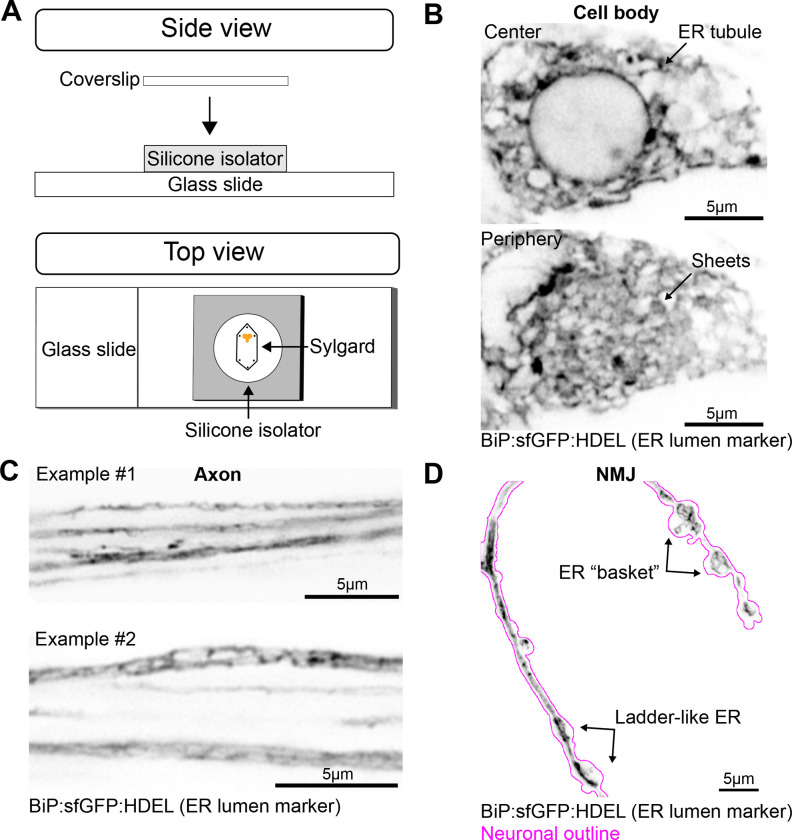
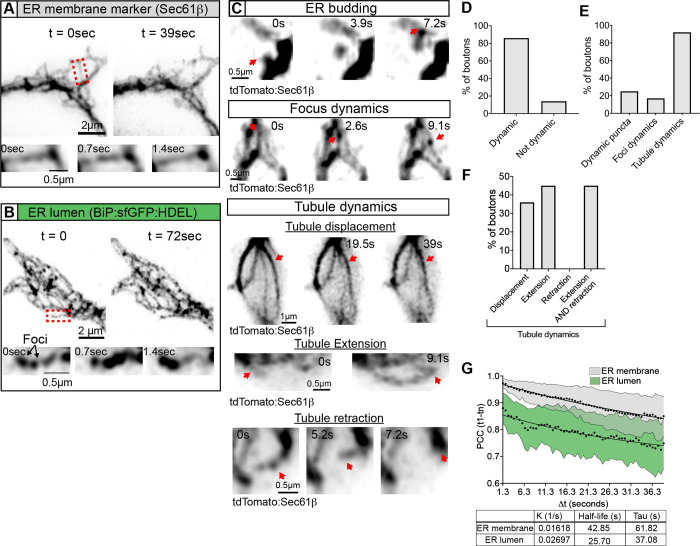
The endoplasmic reticulum (ER) extends throughout neurons and regulates many neuronal functions, including neurite outgrowth, neurotransmission, and synaptic plasticity. Mutations in proteins that control ER shape are linked to the neurodegenerative disorder Hereditary Spastic Paraplegia (HSP), yet the ultrastructure and dynamics of neuronal ER remain largely unexplored, especially at presynaptic terminals. Using super-resolution and live imaging in D. melanogaster larval motor neurons, we investigated ER structure at presynaptic terminals of wild-type animals and null mutants of the ER shaping protein and HSP-linked gene, Atlastin. Previous studies using an ER luminal marker reported diffuse localization at Atlastin mutant presynaptic terminals, which was attributed to ER fragmentation. However, using an ER membrane marker, we discovered that Atlastin mutant ER forms robust networks with only mild defects in structural dynamics, indicating the primary defect is functional rather than architectural. We demonstrate that Atlastin mutants progressively displace overexpressed luminal ER proteins to the cytosol during larval development, specifically at synapses, while these proteins remain correctly localized in cell bodies, axons, and muscles. This synaptic-specific displacement phenotype, previously unreported in non-neuronal cells, emphasizes the importance of studying neurons to understand HSP pathogenesis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


