{"title":"AFsample: improving multimer prediction with AlphaFold using massive sampling.","authors":"Björn Wallner","doi":"10.1093/bioinformatics/btad573","DOIUrl":null,"url":null,"abstract":"<p><strong>Summary: </strong>The AlphaFold2 neural network model has revolutionized structural biology with unprecedented performance. We demonstrate that by stochastically perturbing the neural network by enabling dropout at inference combined with massive sampling, it is possible to improve the quality of the generated models. We generated ∼6000 models per target compared with 25 default for AlphaFold-Multimer, with v1 and v2 multimer network models, with and without templates, and increased the number of recycles within the network. The method was benchmarked in CASP15, and compared with AlphaFold-Multimer v2 it improved the average DockQ from 0.41 to 0.55 using identical input and was ranked at the very top in the protein assembly category when compared with all other groups participating in CASP15. The simplicity of the method should facilitate the adaptation by the field, and the method should be useful for anyone interested in modeling multimeric structures, alternate conformations, or flexible structures.</p><p><strong>Availability and implementation: </strong>AFsample is available online at http://wallnerlab.org/AFsample.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":" ","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10534052/pdf/","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad573","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 3
Abstract
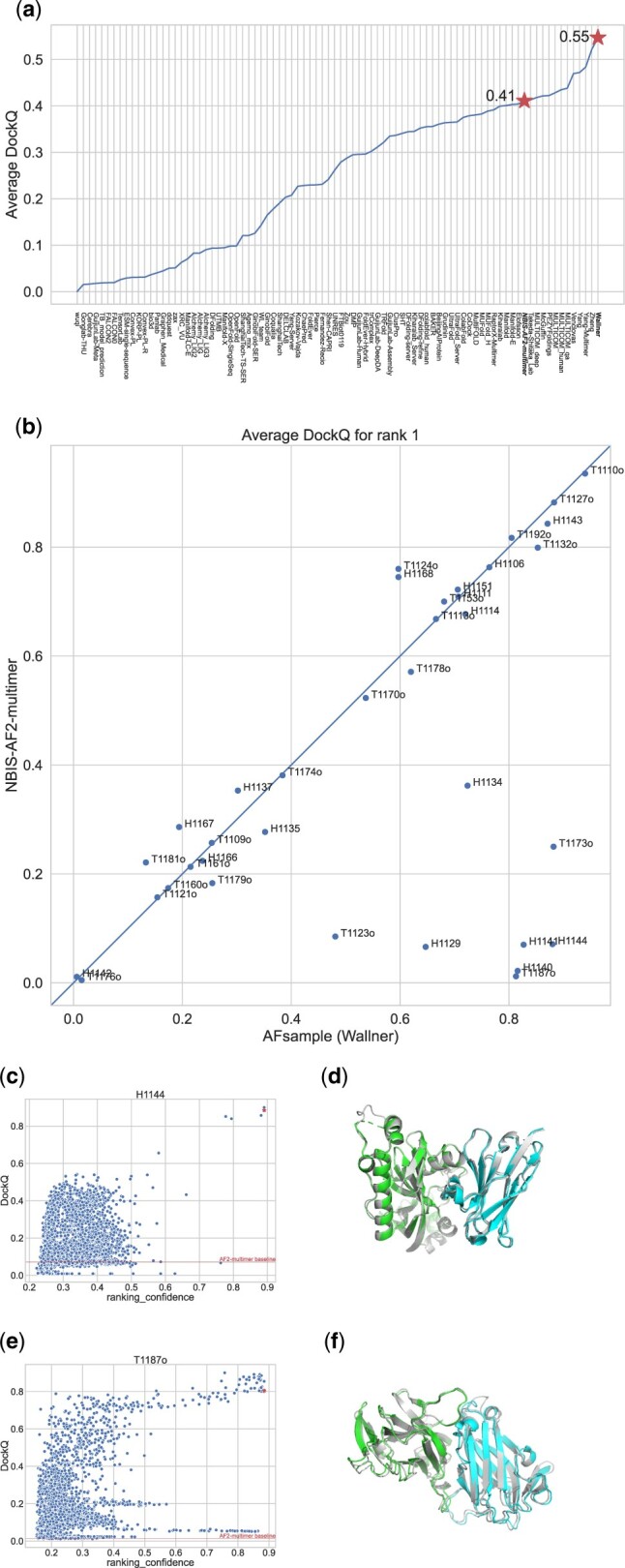
Summary: The AlphaFold2 neural network model has revolutionized structural biology with unprecedented performance. We demonstrate that by stochastically perturbing the neural network by enabling dropout at inference combined with massive sampling, it is possible to improve the quality of the generated models. We generated ∼6000 models per target compared with 25 default for AlphaFold-Multimer, with v1 and v2 multimer network models, with and without templates, and increased the number of recycles within the network. The method was benchmarked in CASP15, and compared with AlphaFold-Multimer v2 it improved the average DockQ from 0.41 to 0.55 using identical input and was ranked at the very top in the protein assembly category when compared with all other groups participating in CASP15. The simplicity of the method should facilitate the adaptation by the field, and the method should be useful for anyone interested in modeling multimeric structures, alternate conformations, or flexible structures.
Availability and implementation: AFsample is available online at http://wallnerlab.org/AFsample.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


