Lea Abou Haidar, Robert C Harris, Panayotis Pachnis, Hongli Chen, Garrett K Gotway, Min Ni, Ralph J DeBerardinis
{"title":"Novel pathogenic <i>UQCRC2</i> variants in a female with normal neurodevelopment.","authors":"Lea Abou Haidar, Robert C Harris, Panayotis Pachnis, Hongli Chen, Garrett K Gotway, Min Ni, Ralph J DeBerardinis","doi":"10.1101/mcs.a006295","DOIUrl":null,"url":null,"abstract":"<p><p>Electron transport chain (ETC) disorders are a group of rare, multisystem diseases caused by impaired oxidative phosphorylation and energy production. Deficiencies in complex III (CIII), also known as ubiquinol-cytochrome <i>c</i> reductase, are particularly rare in humans. Ubiquinol-cytochrome <i>c</i> reductase core protein 2 (<i>UQCRC2</i>) encodes a subunit of CIII that plays a crucial role in dimerization. Several pathogenic <i>UQCRC2</i> variants have been identified in patients presenting with metabolic abnormalities that include lactic acidosis, hyperammonemia, hypoglycemia, and organic aciduria. Almost all previously reported <i>UQCRC2</i>-deficient patients exhibited neurodevelopmental involvement, including developmental delays and structural brain anomalies. Here, we describe a girl who presented at 3 yr of age with lactic acidosis, hyperammonemia, and hypoglycemia but has not shown any evidence of neurodevelopmental dysfunction by age 15. Whole-exome sequencing revealed compound heterozygosity for two novel variants in <i>UQCRC2</i>: c.1189G>A; p.Gly397Arg and c.437T>C; p.Phe146Ser. Here, we discuss the patient's clinical presentation and the likely pathogenicity of these two missense variants.</p>","PeriodicalId":10360,"journal":{"name":"Cold Spring Harbor Molecular Case Studies","volume":" ","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2024-01-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10815277/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cold Spring Harbor Molecular Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006295","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/12/1 0:00:00","PubModel":"Print","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
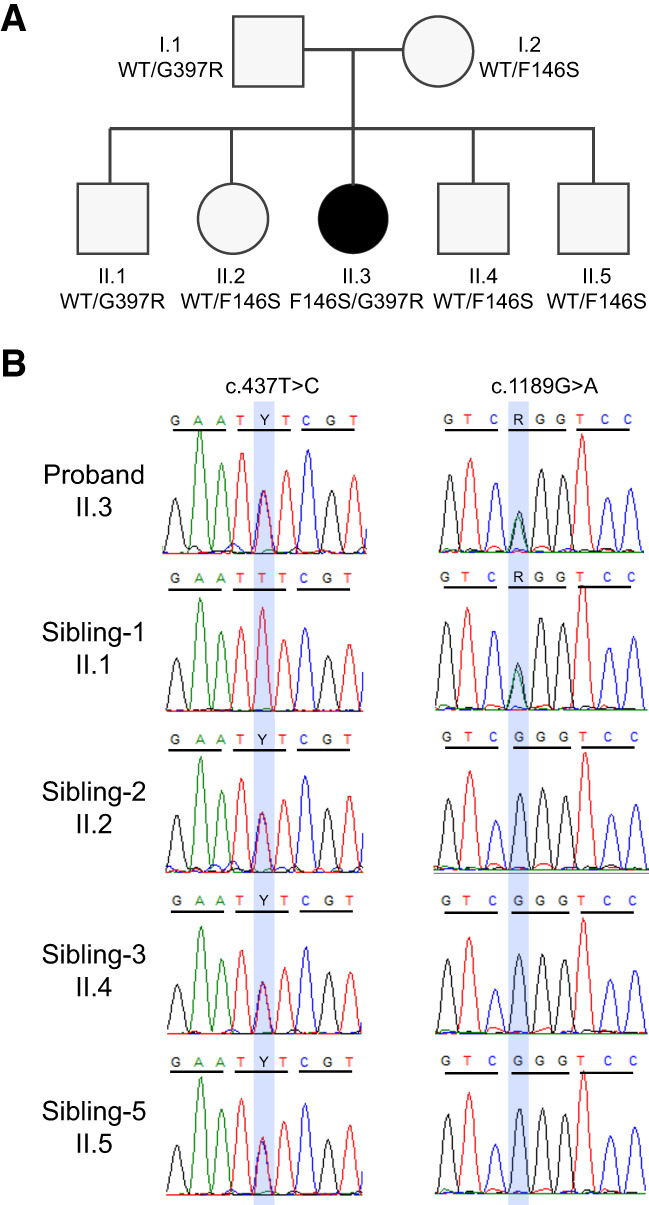
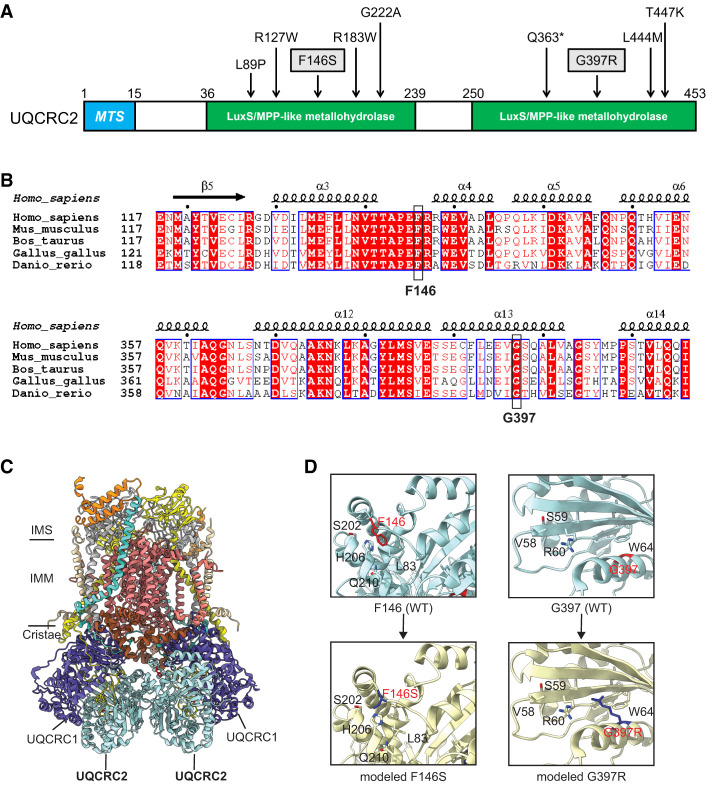
Electron transport chain (ETC) disorders are a group of rare, multisystem diseases caused by impaired oxidative phosphorylation and energy production. Deficiencies in complex III (CIII), also known as ubiquinol-cytochrome c reductase, are particularly rare in humans. Ubiquinol-cytochrome c reductase core protein 2 (UQCRC2) encodes a subunit of CIII that plays a crucial role in dimerization. Several pathogenic UQCRC2 variants have been identified in patients presenting with metabolic abnormalities that include lactic acidosis, hyperammonemia, hypoglycemia, and organic aciduria. Almost all previously reported UQCRC2-deficient patients exhibited neurodevelopmental involvement, including developmental delays and structural brain anomalies. Here, we describe a girl who presented at 3 yr of age with lactic acidosis, hyperammonemia, and hypoglycemia but has not shown any evidence of neurodevelopmental dysfunction by age 15. Whole-exome sequencing revealed compound heterozygosity for two novel variants in UQCRC2: c.1189G>A; p.Gly397Arg and c.437T>C; p.Phe146Ser. Here, we discuss the patient's clinical presentation and the likely pathogenicity of these two missense variants.
期刊介绍:
Cold Spring Harbor Molecular Case Studies is an open-access, peer-reviewed, international journal in the field of precision medicine. Articles in the journal present genomic and molecular analyses of individuals or cohorts alongside their clinical presentations and phenotypic information. The journal''s purpose is to rapidly share insights into disease development and treatment gained by application of genomics, proteomics, metabolomics, biomarker analysis, and other approaches. The journal covers the fields of cancer, complex diseases, monogenic disorders, neurological conditions, orphan diseases, infectious disease, gene therapy, and pharmacogenomics. It has a rapid peer-review process that is based on technical evaluation of the analyses performed, not the novelty of findings, and offers a swift, clear path to publication. The journal publishes: Research Reports presenting detailed case studies of individuals and small cohorts, Research Articles describing more extensive work using larger cohorts and/or functional analyses, Rapid Communications presenting the discovery of a novel variant and/or novel phenotype associated with a known disease gene, Rapid Cancer Communications presenting the discovery of a novel variant or combination of variants in a cancer type, Variant Discrepancy Resolution describing efforts to resolve differences or update variant interpretations in ClinVar through case-level data sharing, Follow-up Reports linked to previous observations, Plus Review Articles, Editorials, and Position Statements on best practices for research in precision medicine.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


