Gherard Batisti Biffignandi, Greta Bellinzona, Greta Petazzoni, Davide Sassera, Gian Vincenzo Zuccotti, Claudio Bandi, Fausto Baldanti, Francesco Comandatore, Stefano Gaiarsa
{"title":"P-DOR, an easy-to-use pipeline to reconstruct bacterial outbreaks using genomics.","authors":"Gherard Batisti Biffignandi, Greta Bellinzona, Greta Petazzoni, Davide Sassera, Gian Vincenzo Zuccotti, Claudio Bandi, Fausto Baldanti, Francesco Comandatore, Stefano Gaiarsa","doi":"10.1093/bioinformatics/btad571","DOIUrl":null,"url":null,"abstract":"<p><strong>Summary: </strong>Bacterial Healthcare-Associated Infections (HAIs) are a major threat worldwide, which can be counteracted by establishing effective infection control measures, guided by constant surveillance and timely epidemiological investigations. Genomics is crucial in modern epidemiology but lacks standard methods and user-friendly software, accessible to users without a strong bioinformatics proficiency. To overcome these issues we developed P-DOR, a novel tool for rapid bacterial outbreak characterization. P-DOR accepts genome assemblies as input, it automatically selects a background of publicly available genomes using k-mer distances and adds it to the analysis dataset before inferring a Single-Nucleotide Polymorphism (SNP)-based phylogeny. Epidemiological clusters are identified considering the phylogenetic tree topology and SNP distances. By analyzing the SNP-distance distribution, the user can gauge the correct threshold. Patient metadata can be inputted as well, to provide a spatio-temporal representation of the outbreak. The entire pipeline is fast and scalable and can be also run on low-end computers.</p><p><strong>Availability and implementation: </strong>P-DOR is implemented in Python3 and R and can be installed using conda environments. It is available from GitHub https://github.com/SteMIDIfactory/P-DOR under the GPL-3.0 license.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":" ","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10533420/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad571","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 1
Abstract
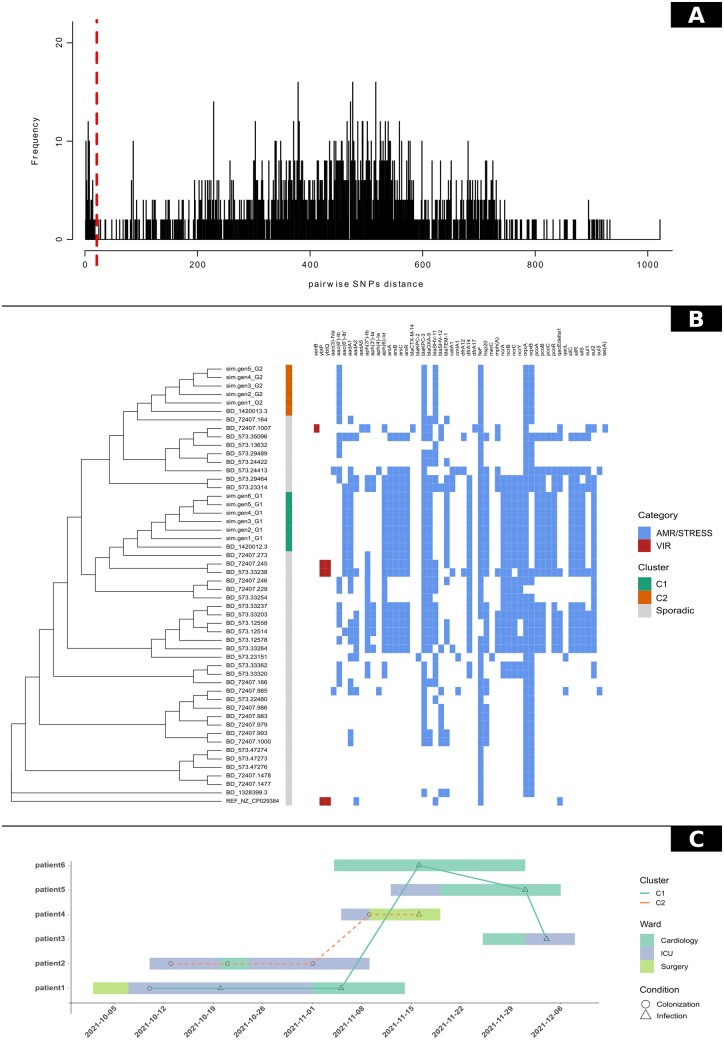
Summary: Bacterial Healthcare-Associated Infections (HAIs) are a major threat worldwide, which can be counteracted by establishing effective infection control measures, guided by constant surveillance and timely epidemiological investigations. Genomics is crucial in modern epidemiology but lacks standard methods and user-friendly software, accessible to users without a strong bioinformatics proficiency. To overcome these issues we developed P-DOR, a novel tool for rapid bacterial outbreak characterization. P-DOR accepts genome assemblies as input, it automatically selects a background of publicly available genomes using k-mer distances and adds it to the analysis dataset before inferring a Single-Nucleotide Polymorphism (SNP)-based phylogeny. Epidemiological clusters are identified considering the phylogenetic tree topology and SNP distances. By analyzing the SNP-distance distribution, the user can gauge the correct threshold. Patient metadata can be inputted as well, to provide a spatio-temporal representation of the outbreak. The entire pipeline is fast and scalable and can be also run on low-end computers.
Availability and implementation: P-DOR is implemented in Python3 and R and can be installed using conda environments. It is available from GitHub https://github.com/SteMIDIfactory/P-DOR under the GPL-3.0 license.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


