Erfan Khorram, Mohammad A Tabatabaiefar, Mehrdad Zeinalian
{"title":"Two Distinct Deleterious Causative Variants in a Family with Multiple Cancer-Affected Patients.","authors":"Erfan Khorram, Mohammad A Tabatabaiefar, Mehrdad Zeinalian","doi":"10.4103/abr.abr_366_22","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Only 5 to 10% of cancers are hereditary, but they are particularly important since they can be passed down from generation to generation, and family members are at elevated risk. Although screening methods are one of the essential strategies for dealing with hereditary cancers, they do not have high specificity and sensitivity. The emergence of whole-exome sequencing (WES) causes a significant increase in the diagnostic rate of cancer-causing variants in at-risk families.</p><p><strong>Materials and methods: </strong>We performed WES on the proband's DNA sample from an Iranian family with multiple cancer-affected members to identify potential causative variants. Multiple in silico tools were used to evaluate the candidate variants' pathogenicity and their effects on the protein's structure, function, and stability. Moreover, the candidate variants were co-segregated in the family with Sanger sequencing.</p><p><strong>Results: </strong>The WES data analysis identified two pathogenic variants (<i>CHEK2:</i> NM_007194.4: c.538C>T, p.Arg180Cys and <i>MLH1:</i> NM_000249.4, c.844G>A, p.Ala282Thr). Sanger sequencing data showed each of the variants was incompletely segregated with phenotype, but both of them explained the patient's phenotype together. Also, the structural analysis demonstrated that due to the variant (c.538C>T), a salt bridge between arginine 180 and glutamic acid 149 was lost. Indeed, several protein stability tools described both variants as destabilizing.</p><p><strong>Conclusion: </strong>Herein, we interestingly identify two distinct deleterious causative variants (<i>CHEK2:</i> NM_007194.4: c.538C>T, p.Arg180Cys and <i>MLH1:</i> NM_000249.4, c.844G>A, p.Ala282Thr) in a family with several cancer-affected members. Furthermore, this study's findings established the utility of WES in the genetic diagnostics of cancer.</p>","PeriodicalId":7225,"journal":{"name":"Advanced Biomedical Research","volume":"12 ","pages":"203"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/cf/89/ABR-12-203.PMC10492615.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advanced Biomedical Research","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4103/abr.abr_366_22","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Only 5 to 10% of cancers are hereditary, but they are particularly important since they can be passed down from generation to generation, and family members are at elevated risk. Although screening methods are one of the essential strategies for dealing with hereditary cancers, they do not have high specificity and sensitivity. The emergence of whole-exome sequencing (WES) causes a significant increase in the diagnostic rate of cancer-causing variants in at-risk families.
Materials and methods: We performed WES on the proband's DNA sample from an Iranian family with multiple cancer-affected members to identify potential causative variants. Multiple in silico tools were used to evaluate the candidate variants' pathogenicity and their effects on the protein's structure, function, and stability. Moreover, the candidate variants were co-segregated in the family with Sanger sequencing.
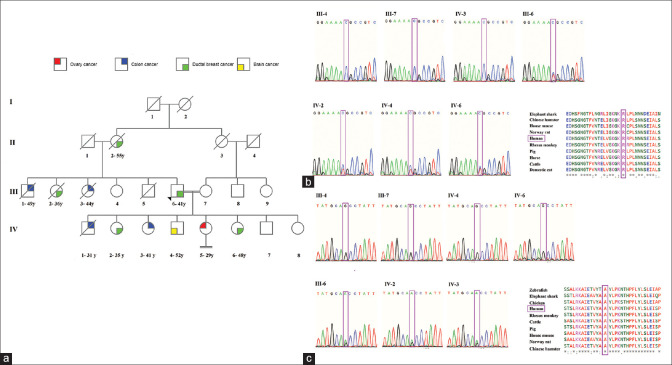
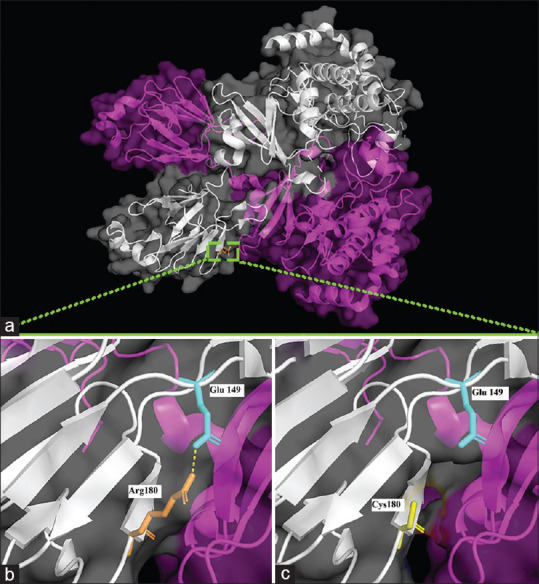
Results: The WES data analysis identified two pathogenic variants (CHEK2: NM_007194.4: c.538C>T, p.Arg180Cys and MLH1: NM_000249.4, c.844G>A, p.Ala282Thr). Sanger sequencing data showed each of the variants was incompletely segregated with phenotype, but both of them explained the patient's phenotype together. Also, the structural analysis demonstrated that due to the variant (c.538C>T), a salt bridge between arginine 180 and glutamic acid 149 was lost. Indeed, several protein stability tools described both variants as destabilizing.
Conclusion: Herein, we interestingly identify two distinct deleterious causative variants (CHEK2: NM_007194.4: c.538C>T, p.Arg180Cys and MLH1: NM_000249.4, c.844G>A, p.Ala282Thr) in a family with several cancer-affected members. Furthermore, this study's findings established the utility of WES in the genetic diagnostics of cancer.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


