Rana German, Natascia Marino, Chris Hemmerich, Ram Podicheti, Douglas B Rusch, Leah T Stiemsma, Hongyu Gao, Xiaoling Xuei, Pam Rockey, Anna Maria Storniolo
{"title":"Exploring breast tissue microbial composition and the association with breast cancer risk factors.","authors":"Rana German, Natascia Marino, Chris Hemmerich, Ram Podicheti, Douglas B Rusch, Leah T Stiemsma, Hongyu Gao, Xiaoling Xuei, Pam Rockey, Anna Maria Storniolo","doi":"10.1186/s13058-023-01677-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Microbial dysbiosis has emerged as an important element in the development and progression of various cancers, including breast cancer. However, the microbial composition of the breast from healthy individuals, even relative to risk of developing breast cancer, remains unclear. Here, we performed a comprehensive analysis of the microbiota of the normal breast tissue, which was analyzed in relation to the microbial composition of the tumor and adjacent normal tissue.</p><p><strong>Methods: </strong>The study cohorts included 403 cancer-free women (who donated normal breast tissue cores) and 76 breast cancer patients (who donated tumor and/or adjacent normal tissue samples). Microbiome profiling was obtained by sequencing the nine hypervariable regions of the 16S rRNA gene (V1V2, V2V3, V3V4, V4V5, V5V7, and V7V9). Transcriptome analysis was also performed on 190 normal breast tissue samples. Breast cancer risk score was assessed using the Tyrer-Cuzick risk model.</p><p><strong>Results: </strong>The V1V2 amplicon sequencing resulted more suitable for the analysis of the normal breast microbiome and identified Lactobacillaceae (Firmicutes phylum), Acetobacterraceae, and Xanthomonadaceae (both Proteobacteria phylum) as the most abundant families in the normal breast. However, Ralstonia (Proteobacteria phylum) was more abundant in both breast tumors and histologically normal tissues adjacent to malignant tumors. We also conducted a correlation analysis between the microbiome and known breast cancer risk factors. Abundances of the bacterial taxa Acetotobacter aceti, Lactobacillus vini, Lactobacillus paracasei, and Xanthonomas sp. were associated with age (p < 0.0001), racial background (p < 0.0001), and parity (p < 0.0001). Finally, transcriptome analysis of normal breast tissues showed an enrichment in metabolism- and immune-related genes in the tissues with abundant Acetotobacter aceti, Lactobacillus vini, Lactobacillus paracasei, and Xanthonomas sp., whereas the presence of Ralstonia in the normal tissue was linked to dysregulation of genes involved in the carbohydrate metabolic pathway.</p><p><strong>Conclusions: </strong>This study defines the microbial features of normal breast tissue, thus providing a basis to understand cancer-related dysbiosis. Moreover, the findings reveal that lifestyle factors can significantly affect the normal breast microbial composition.</p>","PeriodicalId":9283,"journal":{"name":"Breast Cancer Research : BCR","volume":"25 1","pages":"82"},"PeriodicalIF":0.0000,"publicationDate":"2023-07-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10332106/pdf/","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Breast Cancer Research : BCR","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13058-023-01677-6","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 2
Abstract
Background: Microbial dysbiosis has emerged as an important element in the development and progression of various cancers, including breast cancer. However, the microbial composition of the breast from healthy individuals, even relative to risk of developing breast cancer, remains unclear. Here, we performed a comprehensive analysis of the microbiota of the normal breast tissue, which was analyzed in relation to the microbial composition of the tumor and adjacent normal tissue.
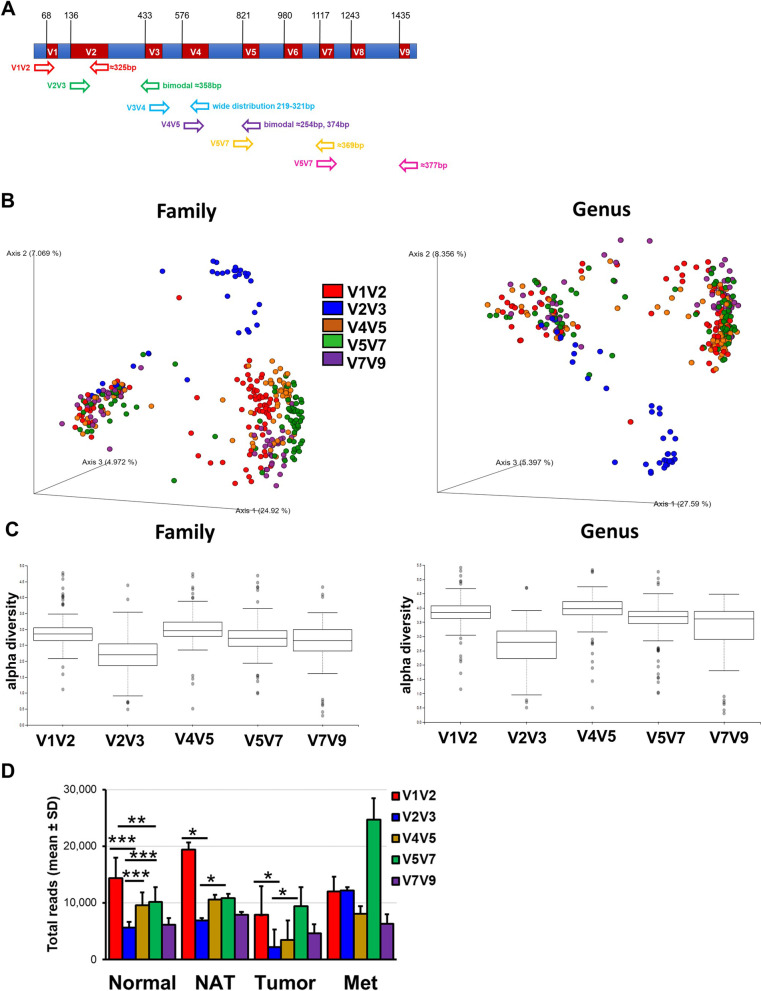
Methods: The study cohorts included 403 cancer-free women (who donated normal breast tissue cores) and 76 breast cancer patients (who donated tumor and/or adjacent normal tissue samples). Microbiome profiling was obtained by sequencing the nine hypervariable regions of the 16S rRNA gene (V1V2, V2V3, V3V4, V4V5, V5V7, and V7V9). Transcriptome analysis was also performed on 190 normal breast tissue samples. Breast cancer risk score was assessed using the Tyrer-Cuzick risk model.
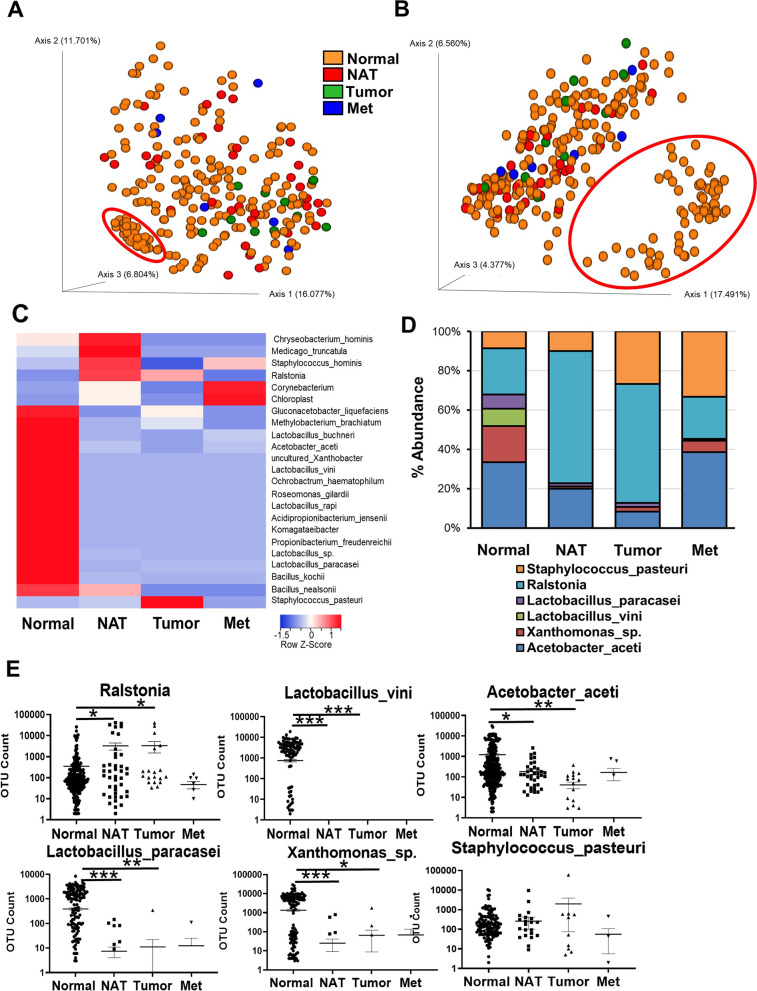
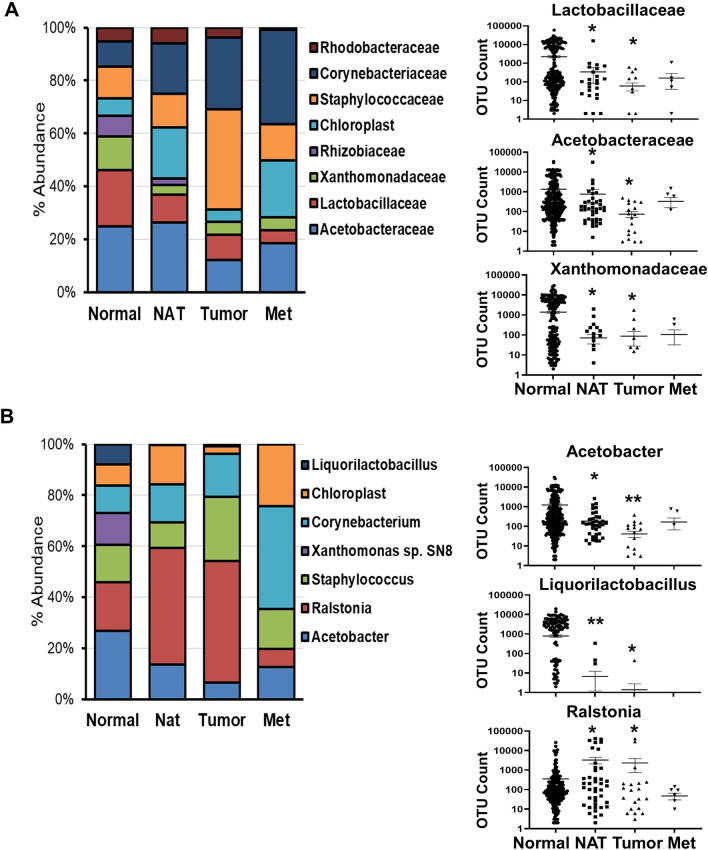
Results: The V1V2 amplicon sequencing resulted more suitable for the analysis of the normal breast microbiome and identified Lactobacillaceae (Firmicutes phylum), Acetobacterraceae, and Xanthomonadaceae (both Proteobacteria phylum) as the most abundant families in the normal breast. However, Ralstonia (Proteobacteria phylum) was more abundant in both breast tumors and histologically normal tissues adjacent to malignant tumors. We also conducted a correlation analysis between the microbiome and known breast cancer risk factors. Abundances of the bacterial taxa Acetotobacter aceti, Lactobacillus vini, Lactobacillus paracasei, and Xanthonomas sp. were associated with age (p < 0.0001), racial background (p < 0.0001), and parity (p < 0.0001). Finally, transcriptome analysis of normal breast tissues showed an enrichment in metabolism- and immune-related genes in the tissues with abundant Acetotobacter aceti, Lactobacillus vini, Lactobacillus paracasei, and Xanthonomas sp., whereas the presence of Ralstonia in the normal tissue was linked to dysregulation of genes involved in the carbohydrate metabolic pathway.
Conclusions: This study defines the microbial features of normal breast tissue, thus providing a basis to understand cancer-related dysbiosis. Moreover, the findings reveal that lifestyle factors can significantly affect the normal breast microbial composition.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


