Hua Feng, Fangyu Wang, Ning Li, Qian Xu, Guanming Zheng, Xuefeng Sun, Man Hu, Xuewu Li, Guangxu Xing, Gaiping Zhang
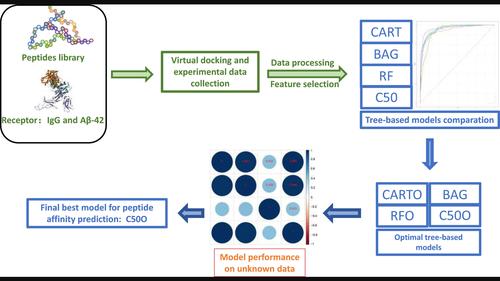
{"title":"Use of tree-based machine learning methods to screen affinitive peptides based on docking data.","authors":"Hua Feng, Fangyu Wang, Ning Li, Qian Xu, Guanming Zheng, Xuefeng Sun, Man Hu, Xuewu Li, Guangxu Xing, Gaiping Zhang","doi":"10.1002/minf.202300143","DOIUrl":null,"url":null,"abstract":"<p><p>Screening peptides with good affinity is an important step in peptide-drug discovery. Recent advancement in computer and data science have made machine learning a useful tool in accurately affinitive-peptide screening. In current study, four different tree-based algorithms, including Classification and regression trees (CART), C5.0 decision tree (C50), Bagged CART (BAG) and Random Forest (RF), were employed to explore the relationship between experimental peptide affinities and virtual docking data, and the performance of each model was also compared in parallel. All four algorithms showed better performances on dataset pre-scaled, -centered and -PCA than other pre-processed dataset. After model re-built and hyperparameter optimization, the optimal C50 model (C50O) showed the best performances in terms of Accuracy, Kappa, Sensitivity, Specificity, F1, MCC and AUC when validated on test data and an unknown PEDV datasets evaluation (Accuracy=80.4 %). BAG and RFO (the optimal RF), as two best models during training process, did not performed as expecting during in testing and unknown dataset validations. Furthermore, the high correlation of the predictions of RFO and BAG to C50O implied the high stability and robustness of their prediction. Whereas although the good performance on unknown dataset, the poor performance in test data validation and correlation analysis indicated CARTO could not be used for future data prediction. To accurately evaluate the peptide affinity, the current study firstly gave a tree-model competition on affinitive peptide prediction by using virtual docking data, which would expand the application of machine learning algorithms in studying PepPIs and benefit the development of peptide therapeutics.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":" ","pages":"e202300143"},"PeriodicalIF":3.1000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202300143","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/11/9 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
Screening peptides with good affinity is an important step in peptide-drug discovery. Recent advancement in computer and data science have made machine learning a useful tool in accurately affinitive-peptide screening. In current study, four different tree-based algorithms, including Classification and regression trees (CART), C5.0 decision tree (C50), Bagged CART (BAG) and Random Forest (RF), were employed to explore the relationship between experimental peptide affinities and virtual docking data, and the performance of each model was also compared in parallel. All four algorithms showed better performances on dataset pre-scaled, -centered and -PCA than other pre-processed dataset. After model re-built and hyperparameter optimization, the optimal C50 model (C50O) showed the best performances in terms of Accuracy, Kappa, Sensitivity, Specificity, F1, MCC and AUC when validated on test data and an unknown PEDV datasets evaluation (Accuracy=80.4 %). BAG and RFO (the optimal RF), as two best models during training process, did not performed as expecting during in testing and unknown dataset validations. Furthermore, the high correlation of the predictions of RFO and BAG to C50O implied the high stability and robustness of their prediction. Whereas although the good performance on unknown dataset, the poor performance in test data validation and correlation analysis indicated CARTO could not be used for future data prediction. To accurately evaluate the peptide affinity, the current study firstly gave a tree-model competition on affinitive peptide prediction by using virtual docking data, which would expand the application of machine learning algorithms in studying PepPIs and benefit the development of peptide therapeutics.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


