{"title":"Automated machine learning for genome wide association studies.","authors":"Kleanthi Lakiotaki, Zaharias Papadovasilakis, Vincenzo Lagani, Stefanos Fafalios, Paulos Charonyktakis, Michail Tsagris, Ioannis Tsamardinos","doi":"10.1093/bioinformatics/btad545","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Genome-wide association studies (GWAS) present several computational and statistical challenges for their data analysis, including knowledge discovery, interpretability, and translation to clinical practice.</p><p><strong>Results: </strong>We develop, apply, and comparatively evaluate an automated machine learning (AutoML) approach, customized for genomic data that delivers reliable predictive and diagnostic models, the set of genetic variants that are important for predictions (called a biosignature), and an estimate of the out-of-sample predictive power. This AutoML approach discovers variants with higher predictive performance compared to standard GWAS methods, computes an individual risk prediction score, generalizes to new, unseen data, is shown to better differentiate causal variants from other highly correlated variants, and enhances knowledge discovery and interpretability by reporting multiple equivalent biosignatures.</p><p><strong>Availability and implementation: </strong>Code for this study is available at: https://github.com/mensxmachina/autoML-GWAS. JADBio offers a free version at: https://jadbio.com/sign-up/. SNP data can be downloaded from the EGA repository (https://ega-archive.org/). PRS data are found at: https://www.aicrowd.com/challenges/opensnp-height-prediction. Simulation data to study population structure can be found at: https://easygwas.ethz.ch/data/public/dataset/view/1/.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":" ","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10562960/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad545","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: Genome-wide association studies (GWAS) present several computational and statistical challenges for their data analysis, including knowledge discovery, interpretability, and translation to clinical practice.
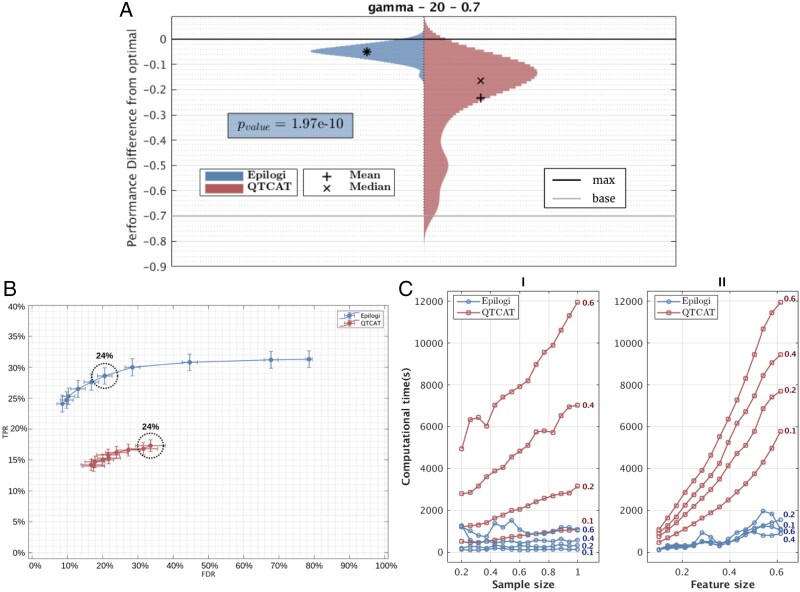
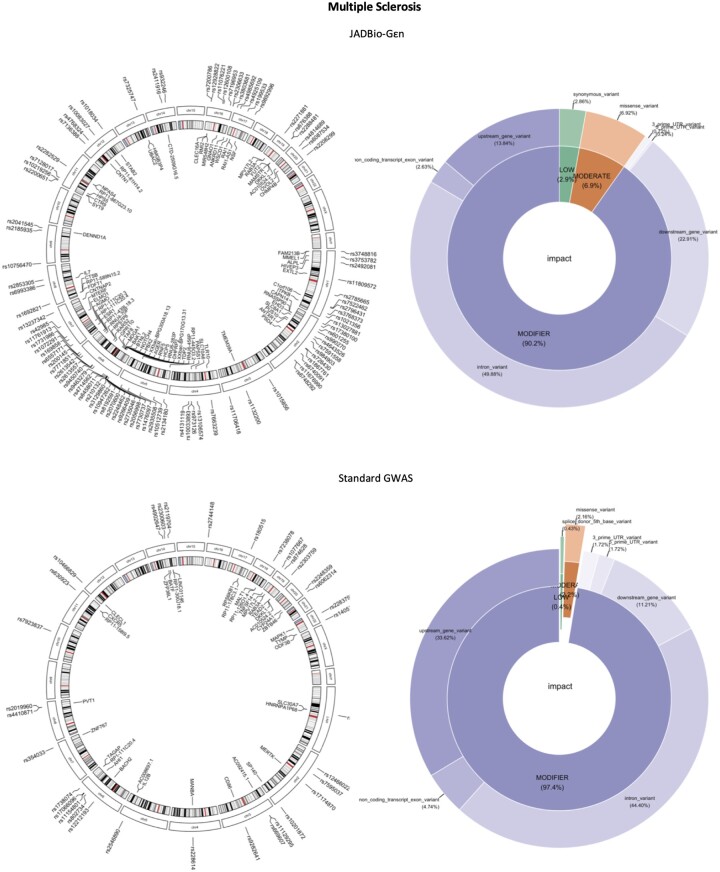
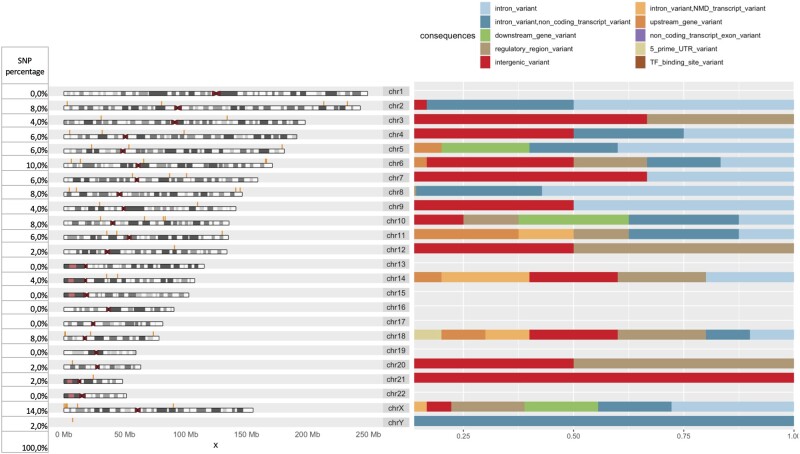
Results: We develop, apply, and comparatively evaluate an automated machine learning (AutoML) approach, customized for genomic data that delivers reliable predictive and diagnostic models, the set of genetic variants that are important for predictions (called a biosignature), and an estimate of the out-of-sample predictive power. This AutoML approach discovers variants with higher predictive performance compared to standard GWAS methods, computes an individual risk prediction score, generalizes to new, unseen data, is shown to better differentiate causal variants from other highly correlated variants, and enhances knowledge discovery and interpretability by reporting multiple equivalent biosignatures.
Availability and implementation: Code for this study is available at: https://github.com/mensxmachina/autoML-GWAS. JADBio offers a free version at: https://jadbio.com/sign-up/. SNP data can be downloaded from the EGA repository (https://ega-archive.org/). PRS data are found at: https://www.aicrowd.com/challenges/opensnp-height-prediction. Simulation data to study population structure can be found at: https://easygwas.ethz.ch/data/public/dataset/view/1/.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


