Madhusudan Rajendran, Maureen C Ferran, Leora Mouli, Gregory A Babbitt, Miranda L Lynch
{"title":"Evolution of drug resistance drives destabilization of flap region dynamics in HIV-1 protease.","authors":"Madhusudan Rajendran, Maureen C Ferran, Leora Mouli, Gregory A Babbitt, Miranda L Lynch","doi":"10.1016/j.bpr.2023.100121","DOIUrl":null,"url":null,"abstract":"<p><p>The HIV-1 protease is one of several common key targets of combination drug therapies for human immunodeficiency virus infection and acquired immunodeficiency syndrome. During the progression of the disease, some individual patients acquire drug resistance due to mutational hotspots on the viral proteins targeted by combination drug therapies. It has recently been discovered that drug-resistant mutations accumulate on the \"flap region\" of the HIV-1 protease, which is a critical dynamic region involved in nonspecific polypeptide binding during invasion and infection of the host cell. In this study, we utilize machine learning-assisted comparative molecular dynamics, conducted at single amino acid site resolution, to investigate the dynamic changes that occur during functional dimerization and drug binding of wild-type and common drug-resistant versions of the main protease. We also use a multiagent machine learning model to identify conserved dynamics of the HIV-1 main protease that are preserved across simian and feline protease orthologs. We find that a key conserved functional site in the flap region, a solvent-exposed isoleucine (Ile50) that controls flap dynamics is functionally targeted by drug resistance mutations, leading to amplified molecular dynamics affecting the functional ability of the flap region to hold the drugs. We conclude that better long-term patient outcomes may be achieved by designing drugs that target protease regions that are less dependent upon single sites with large functional binding effects.</p>","PeriodicalId":72402,"journal":{"name":"Biophysical reports","volume":"3 3","pages":"100121"},"PeriodicalIF":2.7000,"publicationDate":"2023-09-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/6c/83/main.PMC10469570.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biophysical reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.bpr.2023.100121","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOPHYSICS","Score":null,"Total":0}
引用次数: 1
Abstract
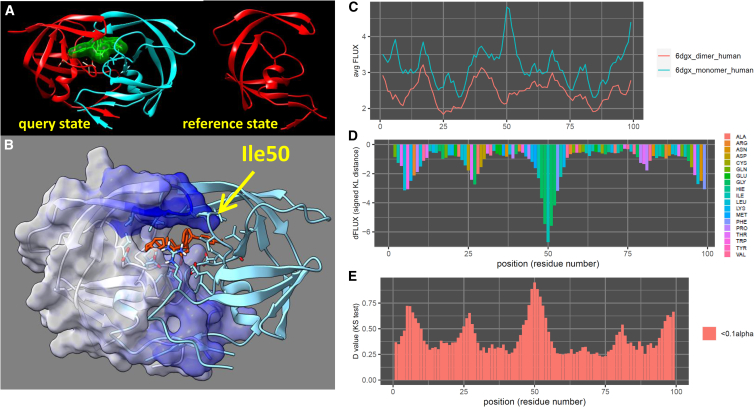
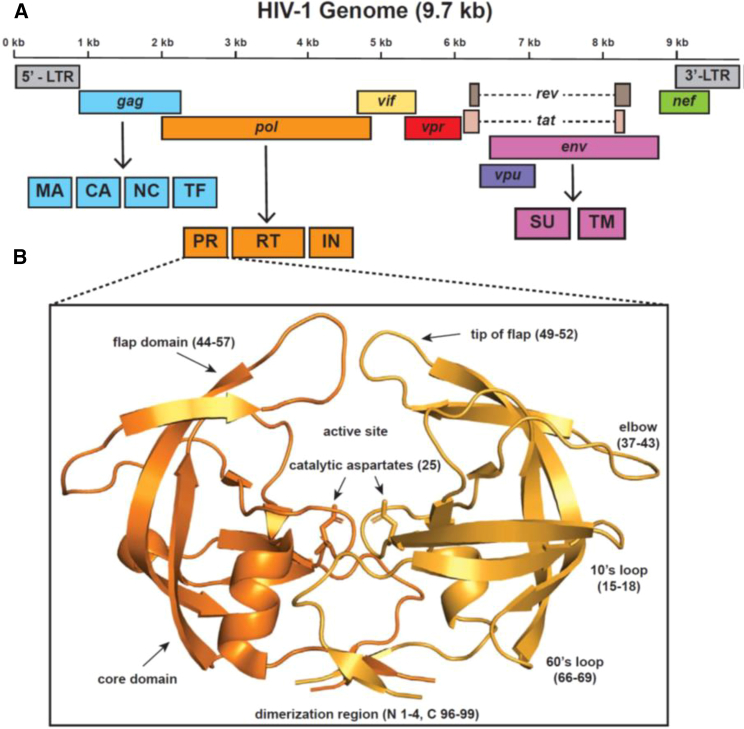
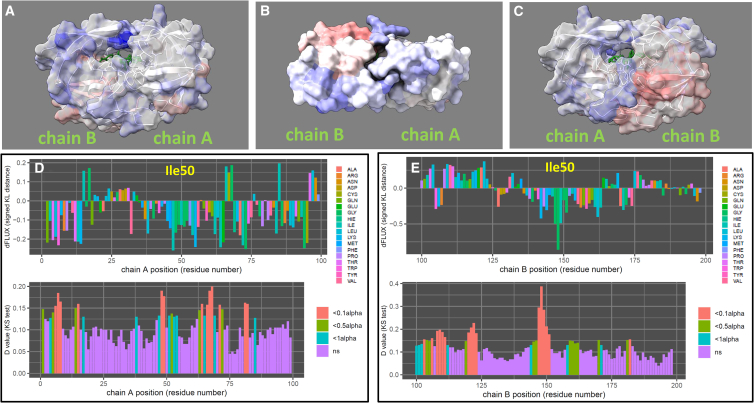
The HIV-1 protease is one of several common key targets of combination drug therapies for human immunodeficiency virus infection and acquired immunodeficiency syndrome. During the progression of the disease, some individual patients acquire drug resistance due to mutational hotspots on the viral proteins targeted by combination drug therapies. It has recently been discovered that drug-resistant mutations accumulate on the "flap region" of the HIV-1 protease, which is a critical dynamic region involved in nonspecific polypeptide binding during invasion and infection of the host cell. In this study, we utilize machine learning-assisted comparative molecular dynamics, conducted at single amino acid site resolution, to investigate the dynamic changes that occur during functional dimerization and drug binding of wild-type and common drug-resistant versions of the main protease. We also use a multiagent machine learning model to identify conserved dynamics of the HIV-1 main protease that are preserved across simian and feline protease orthologs. We find that a key conserved functional site in the flap region, a solvent-exposed isoleucine (Ile50) that controls flap dynamics is functionally targeted by drug resistance mutations, leading to amplified molecular dynamics affecting the functional ability of the flap region to hold the drugs. We conclude that better long-term patient outcomes may be achieved by designing drugs that target protease regions that are less dependent upon single sites with large functional binding effects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


