Tatiana Burrinha, César Cunha, Michael J Hall, Mafalda Lopes-da-Silva, Miguel C Seabra, Cláudia Guimas Almeida
{"title":"Deacidification of endolysosomes by neuronal aging drives synapse loss.","authors":"Tatiana Burrinha, César Cunha, Michael J Hall, Mafalda Lopes-da-Silva, Miguel C Seabra, Cláudia Guimas Almeida","doi":"10.1111/tra.12889","DOIUrl":null,"url":null,"abstract":"<p><p>Previously, we found that age-dependent accumulation of beta-amyloid is not sufficient to cause synaptic decline. Late-endocytic organelles (LEOs) may be driving synaptic decline as lysosomes (Lys) are a target of cellular aging and relevant for synapses. We found that LAMP1-positive LEOs increased in size and number and accumulated near synapses in aged neurons and brains. LEOs' distal accumulation might relate to the increased anterograde movement in aged neurons. Dissecting the LEOs, we found that late-endosomes accumulated while there are fewer terminal Lys in aged neurites, but not in the cell body. The most abundant LEOs were degradative Lys or endolysosomes (ELys), especially in neurites. ELys activity was reduced because of acidification defects, supported by the reduction in v-ATPase subunit V0a1 with aging. Increasing the acidification of aged ELys recovered degradation and reverted synaptic decline, while alkalinization or v-ATPase inhibition, mimicked age-dependent Lys and synapse dysfunction. We identify ELys deacidification as a neuronal mechanism of age-dependent synapse loss. Our findings suggest that future therapeutic strategies to address endolysosomal defects might be able to delay age-related synaptic decline.</p>","PeriodicalId":23207,"journal":{"name":"Traffic","volume":"24 8","pages":"334-354"},"PeriodicalIF":2.5000,"publicationDate":"2023-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Traffic","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1111/tra.12889","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 2
Abstract
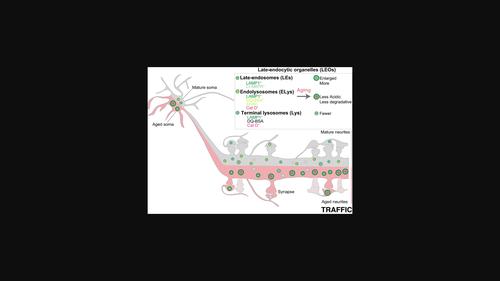
Previously, we found that age-dependent accumulation of beta-amyloid is not sufficient to cause synaptic decline. Late-endocytic organelles (LEOs) may be driving synaptic decline as lysosomes (Lys) are a target of cellular aging and relevant for synapses. We found that LAMP1-positive LEOs increased in size and number and accumulated near synapses in aged neurons and brains. LEOs' distal accumulation might relate to the increased anterograde movement in aged neurons. Dissecting the LEOs, we found that late-endosomes accumulated while there are fewer terminal Lys in aged neurites, but not in the cell body. The most abundant LEOs were degradative Lys or endolysosomes (ELys), especially in neurites. ELys activity was reduced because of acidification defects, supported by the reduction in v-ATPase subunit V0a1 with aging. Increasing the acidification of aged ELys recovered degradation and reverted synaptic decline, while alkalinization or v-ATPase inhibition, mimicked age-dependent Lys and synapse dysfunction. We identify ELys deacidification as a neuronal mechanism of age-dependent synapse loss. Our findings suggest that future therapeutic strategies to address endolysosomal defects might be able to delay age-related synaptic decline.
期刊介绍:
Traffic encourages and facilitates the publication of papers in any field relating to intracellular transport in health and disease. Traffic papers span disciplines such as developmental biology, neuroscience, innate and adaptive immunity, epithelial cell biology, intracellular pathogens and host-pathogen interactions, among others using any eukaryotic model system. Areas of particular interest include protein, nucleic acid and lipid traffic, molecular motors, intracellular pathogens, intracellular proteolysis, nuclear import and export, cytokinesis and the cell cycle, the interface between signaling and trafficking or localization, protein translocation, the cell biology of adaptive an innate immunity, organelle biogenesis, metabolism, cell polarity and organization, and organelle movement.
All aspects of the structural, molecular biology, biochemistry, genetics, morphology, intracellular signaling and relationship to hereditary or infectious diseases will be covered. Manuscripts must provide a clear conceptual or mechanistic advance. The editors will reject papers that require major changes, including addition of significant experimental data or other significant revision.
Traffic will consider manuscripts of any length, but encourages authors to limit their papers to 16 typeset pages or less.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


