{"title":"A unique case of childhood hypophosphatasia caused by a novel heterozygous 51-bp in-frame deletion in the <i>ALPL</i> gene.","authors":"Kanako Tachikawa, Miwa Yamazaki, Toshimi Michigami","doi":"10.1297/cpe.2023-0019","DOIUrl":null,"url":null,"abstract":"<p><p>Hypophosphatasia (HPP) is caused by inactivating variants of the <i>ALPL</i> gene, which encodes tissue non-specific alkaline phosphatase (TNSALP). Among the six subtypes of HPP, childhood HPP presents after 6 months and before 18 yr of age, and is inherited in both autosomal dominant and autosomal recessive manners. Patients with childhood HPP have variable symptoms, including rickets-like bone changes, low bone mineral density (BMD), short stature, muscle weakness, craniosynostosis, and premature loss of deciduous teeth. Here, we describe a 7-yr-old boy with childhood HPP who showed short stature, impaired ossification of the carpal bones, and low BMD. Genetic testing identified a novel heterozygous 51-bp in-frame deletion in the <i>ALPL</i> gene (c.1482_1532del51), leading to the lack of 17 amino acids between Gly495 and Leu511 (p.Gly495_Leu511del). <i>In vitro</i> transfection experiments revealed the loss of enzymatic activity and the dominant-negative effect of the TNSALP[p.Gly495_Leu511del] variant; thus, the patient was diagnosed as having autosomal dominant HPP. The TNSALP[p.Gly495_Leu511del] variant was localized to the plasma membrane as was the wild-type TNSALP (TNSALP[WT]): however, co-immunoprecipitation experiments suggested a reduced dimerization between TNSALP[p.Gly495_Leu511del] and TNSALP[WT]. This case expands the variable clinical manifestation of childhood HPP and sheds light on the molecular bases underlying the dominant-negative effects of some TNSALP variants.</p>","PeriodicalId":10678,"journal":{"name":"Clinical Pediatric Endocrinology","volume":null,"pages":null},"PeriodicalIF":1.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/12/cf/cpe-32-180.PMC10288296.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2023-0019","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
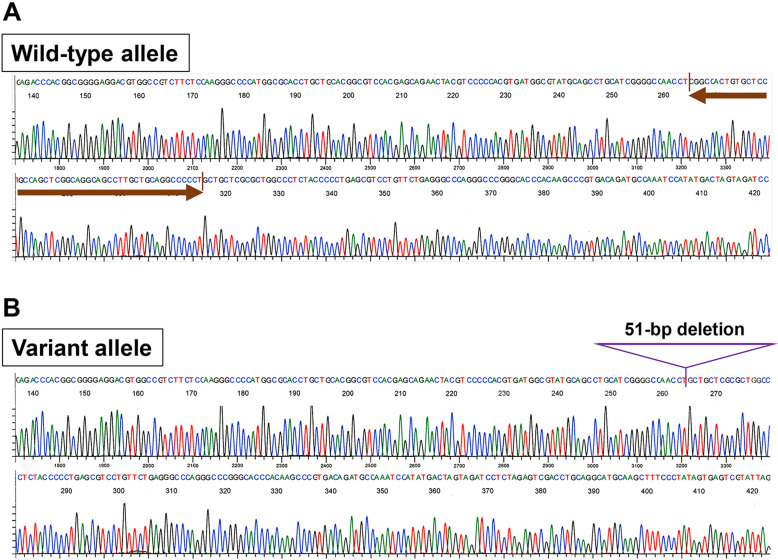
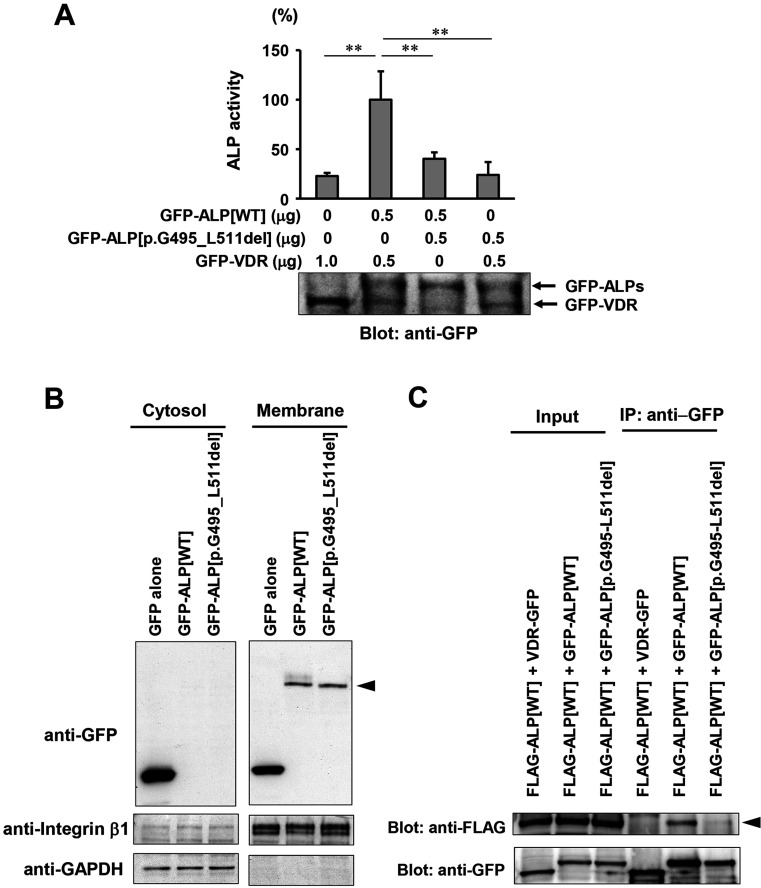
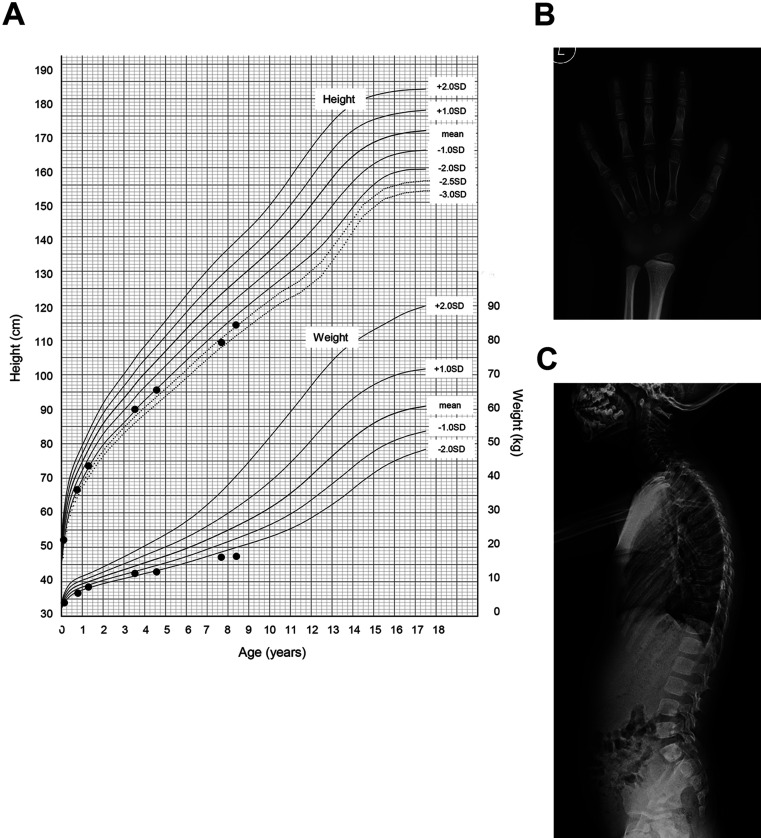
Hypophosphatasia (HPP) is caused by inactivating variants of the ALPL gene, which encodes tissue non-specific alkaline phosphatase (TNSALP). Among the six subtypes of HPP, childhood HPP presents after 6 months and before 18 yr of age, and is inherited in both autosomal dominant and autosomal recessive manners. Patients with childhood HPP have variable symptoms, including rickets-like bone changes, low bone mineral density (BMD), short stature, muscle weakness, craniosynostosis, and premature loss of deciduous teeth. Here, we describe a 7-yr-old boy with childhood HPP who showed short stature, impaired ossification of the carpal bones, and low BMD. Genetic testing identified a novel heterozygous 51-bp in-frame deletion in the ALPL gene (c.1482_1532del51), leading to the lack of 17 amino acids between Gly495 and Leu511 (p.Gly495_Leu511del). In vitro transfection experiments revealed the loss of enzymatic activity and the dominant-negative effect of the TNSALP[p.Gly495_Leu511del] variant; thus, the patient was diagnosed as having autosomal dominant HPP. The TNSALP[p.Gly495_Leu511del] variant was localized to the plasma membrane as was the wild-type TNSALP (TNSALP[WT]): however, co-immunoprecipitation experiments suggested a reduced dimerization between TNSALP[p.Gly495_Leu511del] and TNSALP[WT]. This case expands the variable clinical manifestation of childhood HPP and sheds light on the molecular bases underlying the dominant-negative effects of some TNSALP variants.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


