As a Potential Therapeutic Target, C1q Induces Synapse Loss Via Inflammasome-activating Apoptotic and Mitochondria Impairment Mechanisms in Alzheimer's Disease.
{"title":"As a Potential Therapeutic Target, C1q Induces Synapse Loss Via Inflammasome-activating Apoptotic and Mitochondria Impairment Mechanisms in Alzheimer's Disease.","authors":"Pei-Pei Guan, Tong-Qi Ge, Pu Wang","doi":"10.1007/s11481-023-10076-9","DOIUrl":null,"url":null,"abstract":"<p><p>C1q, the initiator of the classical pathway of the complement system, is activated during Alzheimer's disease (AD) development and progression and is especially associated with the production and deposition of β-amyloid protein (Aβ) and phosphorylated tau in β-amyloid plaques (APs) and neurofibrillary tangles (NFTs). Activation of C1q is responsible for induction of synapse loss, leading to neurodegeneration in AD. Mechanistically, C1q could activate glial cells, which results in the loss of synapses via regulation of synapse pruning and phagocytosis in AD. In addition, C1q induces neuroinflammation by inducing proinflammatory cytokine secretion, which is partially mediated by inflammasome activation. Activation of inflammasomes might mediate the effects of C1q on induction of synapse apoptosis. On the other hand, activation of C1q impairs mitochondria, which hinders the renovation and regeneration of synapses. All these actions of C1q contribute to the loss of synapses during neurodegeneration in AD. Therefore, pharmacological, or genetic interventions targeting C1q may provide potential therapeutic strategies for combating AD.</p>","PeriodicalId":73858,"journal":{"name":"Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology","volume":" ","pages":"267-284"},"PeriodicalIF":6.2000,"publicationDate":"2023-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s11481-023-10076-9","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/6/29 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
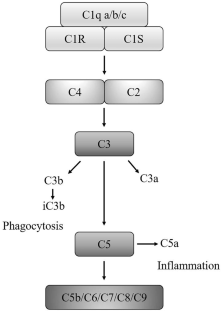
C1q, the initiator of the classical pathway of the complement system, is activated during Alzheimer's disease (AD) development and progression and is especially associated with the production and deposition of β-amyloid protein (Aβ) and phosphorylated tau in β-amyloid plaques (APs) and neurofibrillary tangles (NFTs). Activation of C1q is responsible for induction of synapse loss, leading to neurodegeneration in AD. Mechanistically, C1q could activate glial cells, which results in the loss of synapses via regulation of synapse pruning and phagocytosis in AD. In addition, C1q induces neuroinflammation by inducing proinflammatory cytokine secretion, which is partially mediated by inflammasome activation. Activation of inflammasomes might mediate the effects of C1q on induction of synapse apoptosis. On the other hand, activation of C1q impairs mitochondria, which hinders the renovation and regeneration of synapses. All these actions of C1q contribute to the loss of synapses during neurodegeneration in AD. Therefore, pharmacological, or genetic interventions targeting C1q may provide potential therapeutic strategies for combating AD.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


