Zeyu Zhu, Wenzhe Hou, Yuwen Cao, Haoran Zheng, Wotu Tian, Li Cao
{"title":"新型CAPN1突变引起的76型痉挛性截瘫:3例病例报告及文献复习。","authors":"Zeyu Zhu, Wenzhe Hou, Yuwen Cao, Haoran Zheng, Wotu Tian, Li Cao","doi":"10.1007/s10048-023-00726-8","DOIUrl":null,"url":null,"abstract":"<p><p>Spastic paraplegia type 76 (SPG76) is a subtype of hereditary spastic paraplegia (HSP) caused by calpain-1 (CAPN1) mutations. Our study described the phenotypic and genetic characteristics of three families with spastic ataxia due to various CAPN1 mutations and further explored the pathogenesis of the two novel mutations. The three patients were 48, 39, and 48 years old, respectively. Patients 1 and 3 were from consanguineous families, while patient 2 was sporadic. Physical examination showed hypertonia, hyperreflexia, and Babinski signs in the lower limbs. Patients 2 and 3 additionally had dysarthria and depression. CAPN1 mutations were identified by whole-exome sequencing, followed by Sanger sequencing and co-segregation analysis within the family. Functional examination of the newly identified mutations was further explored. Two homozygous mutations were detected in patient 1 (c.213dupG, p.D72Gfs*95) and patient 3 (c.1729+1G>A) with HSP, respectively. Patient 2 had compound heterozygous mutations c.853C>T (p.R285X) and c.1324G>A (p.G442S). Western blotting revealed the p.D72Gfs*95 with a smaller molecular weight than WT and p.G442S. In vitro, the wild-type calpain-1 is mostly located in the cytoplasm and colocalized with tubulin by immunostaining. However, p.D72Gfs*95 and p.G442S abnormally formed intracellular aggregation, with little colocalization with tubulin. In this study, we identified three cases with SPG76, due to four various CAPN1 mutations, presenting lower limb spasticity and ataxia, with or without bulbar involvement and emotional disorder. Among these, c.213dupG and c.1324G>A are first identified in this paper. The genotype-phenotype correlation of the SPG76 cases reported worldwide was further summarized.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":" ","pages":"243-250"},"PeriodicalIF":1.6000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Spastic paraplegia type 76 due to novel CAPN1 mutations: three case reports with literature review.\",\"authors\":\"Zeyu Zhu, Wenzhe Hou, Yuwen Cao, Haoran Zheng, Wotu Tian, Li Cao\",\"doi\":\"10.1007/s10048-023-00726-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Spastic paraplegia type 76 (SPG76) is a subtype of hereditary spastic paraplegia (HSP) caused by calpain-1 (CAPN1) mutations. Our study described the phenotypic and genetic characteristics of three families with spastic ataxia due to various CAPN1 mutations and further explored the pathogenesis of the two novel mutations. The three patients were 48, 39, and 48 years old, respectively. Patients 1 and 3 were from consanguineous families, while patient 2 was sporadic. Physical examination showed hypertonia, hyperreflexia, and Babinski signs in the lower limbs. Patients 2 and 3 additionally had dysarthria and depression. CAPN1 mutations were identified by whole-exome sequencing, followed by Sanger sequencing and co-segregation analysis within the family. Functional examination of the newly identified mutations was further explored. Two homozygous mutations were detected in patient 1 (c.213dupG, p.D72Gfs*95) and patient 3 (c.1729+1G>A) with HSP, respectively. Patient 2 had compound heterozygous mutations c.853C>T (p.R285X) and c.1324G>A (p.G442S). Western blotting revealed the p.D72Gfs*95 with a smaller molecular weight than WT and p.G442S. In vitro, the wild-type calpain-1 is mostly located in the cytoplasm and colocalized with tubulin by immunostaining. However, p.D72Gfs*95 and p.G442S abnormally formed intracellular aggregation, with little colocalization with tubulin. In this study, we identified three cases with SPG76, due to four various CAPN1 mutations, presenting lower limb spasticity and ataxia, with or without bulbar involvement and emotional disorder. Among these, c.213dupG and c.1324G>A are first identified in this paper. The genotype-phenotype correlation of the SPG76 cases reported worldwide was further summarized.</p>\",\"PeriodicalId\":56106,\"journal\":{\"name\":\"Neurogenetics\",\"volume\":\" \",\"pages\":\"243-250\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2023-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10048-023-00726-8\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/7/19 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00726-8","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/7/19 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Spastic paraplegia type 76 due to novel CAPN1 mutations: three case reports with literature review.
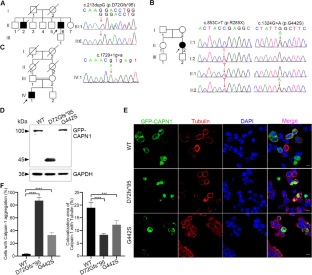
Spastic paraplegia type 76 (SPG76) is a subtype of hereditary spastic paraplegia (HSP) caused by calpain-1 (CAPN1) mutations. Our study described the phenotypic and genetic characteristics of three families with spastic ataxia due to various CAPN1 mutations and further explored the pathogenesis of the two novel mutations. The three patients were 48, 39, and 48 years old, respectively. Patients 1 and 3 were from consanguineous families, while patient 2 was sporadic. Physical examination showed hypertonia, hyperreflexia, and Babinski signs in the lower limbs. Patients 2 and 3 additionally had dysarthria and depression. CAPN1 mutations were identified by whole-exome sequencing, followed by Sanger sequencing and co-segregation analysis within the family. Functional examination of the newly identified mutations was further explored. Two homozygous mutations were detected in patient 1 (c.213dupG, p.D72Gfs*95) and patient 3 (c.1729+1G>A) with HSP, respectively. Patient 2 had compound heterozygous mutations c.853C>T (p.R285X) and c.1324G>A (p.G442S). Western blotting revealed the p.D72Gfs*95 with a smaller molecular weight than WT and p.G442S. In vitro, the wild-type calpain-1 is mostly located in the cytoplasm and colocalized with tubulin by immunostaining. However, p.D72Gfs*95 and p.G442S abnormally formed intracellular aggregation, with little colocalization with tubulin. In this study, we identified three cases with SPG76, due to four various CAPN1 mutations, presenting lower limb spasticity and ataxia, with or without bulbar involvement and emotional disorder. Among these, c.213dupG and c.1324G>A are first identified in this paper. The genotype-phenotype correlation of the SPG76 cases reported worldwide was further summarized.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


