Waqas Amber Gill, Muhammad Usman Khan, Zunaira Shafiq, Muhammad Ramzan Saeed Ashraf Janjua
{"title":"探索二硫化碳二聚体中的分子间相互作用:利用改进的Lennard–Jones势能面进行的从头算研究","authors":"Waqas Amber Gill, Muhammad Usman Khan, Zunaira Shafiq, Muhammad Ramzan Saeed Ashraf Janjua","doi":"10.1002/poc.4548","DOIUrl":null,"url":null,"abstract":"<p>Carbon disulfide dimer (CS<sub>2</sub>)<sub>2</sub> is a model system that has been widely studied in the field of computational chemistry because of its relevance to a variety of chemical and biological processes. The (CS<sub>2</sub>)<sub>2</sub> dimer is a relatively simple molecular system composed of two carbon disulfide (CS<sub>2</sub>) molecules interacting with each other through intermolecular forces. Despite its apparent simplicity, the (CS<sub>2</sub>)<sub>2</sub> dimer exhibits a rich array of structural and dynamical properties that are of great interest to researchers. In this research, we present an ab initio study of the intermolecular interactions in the carbon disulfide dimer (CS<sub>2</sub>)<sub>2</sub> using an improved Lennard–Jones (ILJ) potential with CCSD(T)/QZVPP calculations. The potential energy surface of (CS<sub>2</sub>)<sub>2</sub> is calculated using high-level quantum mechanical calculations based on the CCSD(T)/def2-qzvpp method, which accurately accounts for electron correlation effects. The resulting potential energy surface is then fitted to an ILJ potential energy function, which includes both long-range dipole–dipole interactions and short-range repulsive interactions. The calculated potential energy surface reveals a rich variety of structural and dynamical properties of (CS<sub>2</sub>)<sub>2</sub>, including multiple minima and saddle points, which are sensitive to the relative orientation of the two CS<sub>2</sub> molecules. It is essential to use extended basis sets to accurately incorporate the significant quadrupole moment of CS<sub>2</sub>, which we have calculated to be 2.44 a.u. The results of this study demonstrate the importance of using high-level ab initio methods for the accurate calculation of potential energy surfaces in complex molecular systems such as (CS<sub>2</sub>)<sub>2</sub>. The use of an ILJ potential, which takes into account both dipole–dipole interactions and short-range repulsive interactions, provides a more accurate and efficient approach for modeling intermolecular interactions in (CS<sub>2</sub>)<sub>2</sub> and other similar systems. The results of this study will be useful for understanding the behavior of carbon disulfide dimers in different environments and for the development of new materials and chemical processes.</p>","PeriodicalId":16829,"journal":{"name":"Journal of Physical Organic Chemistry","volume":null,"pages":null},"PeriodicalIF":1.9000,"publicationDate":"2023-06-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"RETRACTED:Exploring the intermolecular interactions in carbon disulfide dimer: An ab initio study using an improved Lennard–Jones potential energy surface for physical insights\",\"authors\":\"Waqas Amber Gill, Muhammad Usman Khan, Zunaira Shafiq, Muhammad Ramzan Saeed Ashraf Janjua\",\"doi\":\"10.1002/poc.4548\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Carbon disulfide dimer (CS<sub>2</sub>)<sub>2</sub> is a model system that has been widely studied in the field of computational chemistry because of its relevance to a variety of chemical and biological processes. The (CS<sub>2</sub>)<sub>2</sub> dimer is a relatively simple molecular system composed of two carbon disulfide (CS<sub>2</sub>) molecules interacting with each other through intermolecular forces. Despite its apparent simplicity, the (CS<sub>2</sub>)<sub>2</sub> dimer exhibits a rich array of structural and dynamical properties that are of great interest to researchers. In this research, we present an ab initio study of the intermolecular interactions in the carbon disulfide dimer (CS<sub>2</sub>)<sub>2</sub> using an improved Lennard–Jones (ILJ) potential with CCSD(T)/QZVPP calculations. The potential energy surface of (CS<sub>2</sub>)<sub>2</sub> is calculated using high-level quantum mechanical calculations based on the CCSD(T)/def2-qzvpp method, which accurately accounts for electron correlation effects. The resulting potential energy surface is then fitted to an ILJ potential energy function, which includes both long-range dipole–dipole interactions and short-range repulsive interactions. The calculated potential energy surface reveals a rich variety of structural and dynamical properties of (CS<sub>2</sub>)<sub>2</sub>, including multiple minima and saddle points, which are sensitive to the relative orientation of the two CS<sub>2</sub> molecules. It is essential to use extended basis sets to accurately incorporate the significant quadrupole moment of CS<sub>2</sub>, which we have calculated to be 2.44 a.u. The results of this study demonstrate the importance of using high-level ab initio methods for the accurate calculation of potential energy surfaces in complex molecular systems such as (CS<sub>2</sub>)<sub>2</sub>. The use of an ILJ potential, which takes into account both dipole–dipole interactions and short-range repulsive interactions, provides a more accurate and efficient approach for modeling intermolecular interactions in (CS<sub>2</sub>)<sub>2</sub> and other similar systems. The results of this study will be useful for understanding the behavior of carbon disulfide dimers in different environments and for the development of new materials and chemical processes.</p>\",\"PeriodicalId\":16829,\"journal\":{\"name\":\"Journal of Physical Organic Chemistry\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2023-06-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Physical Organic Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/poc.4548\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, ORGANIC\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physical Organic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/poc.4548","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
RETRACTED:Exploring the intermolecular interactions in carbon disulfide dimer: An ab initio study using an improved Lennard–Jones potential energy surface for physical insights
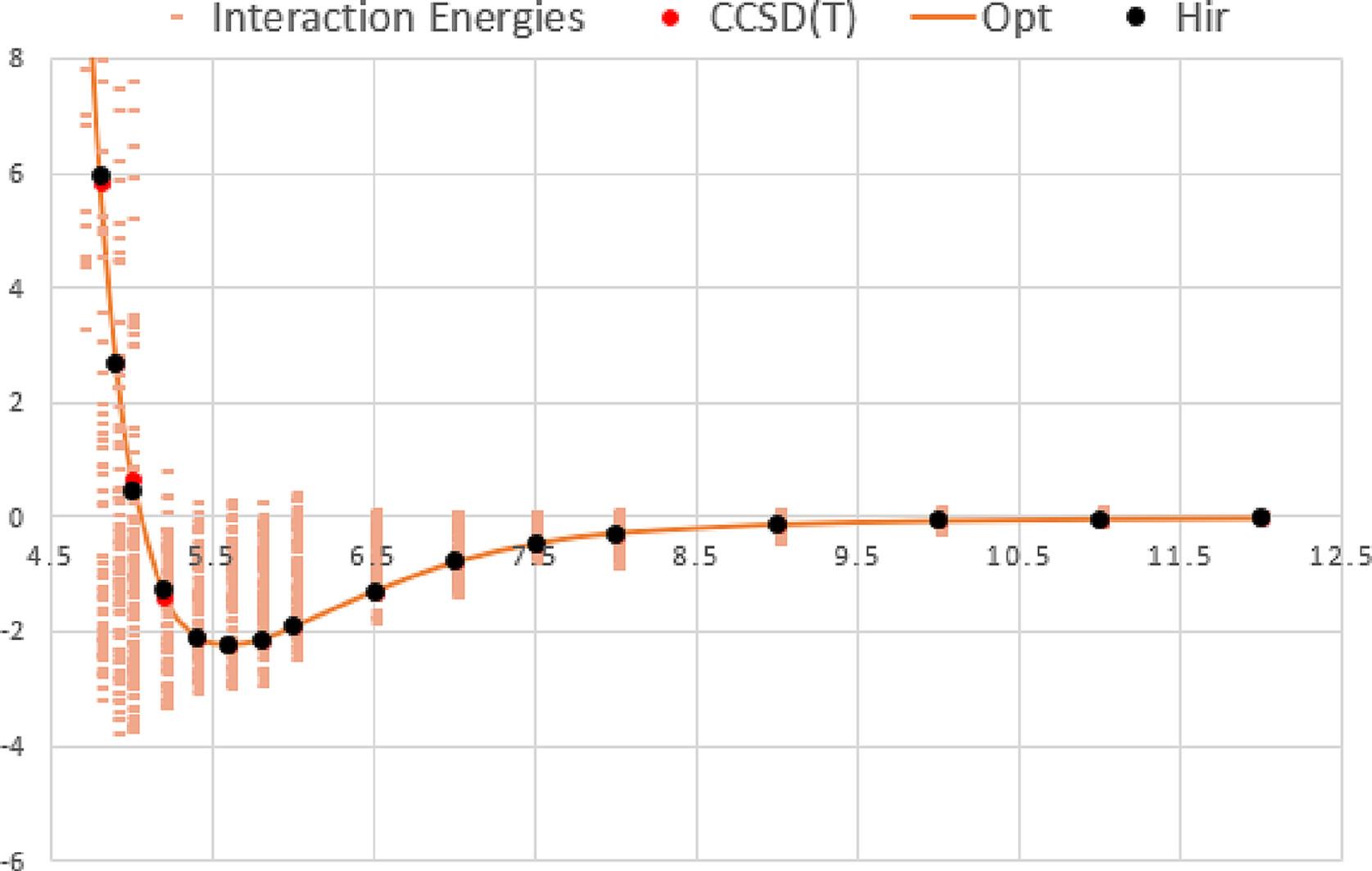
Carbon disulfide dimer (CS2)2 is a model system that has been widely studied in the field of computational chemistry because of its relevance to a variety of chemical and biological processes. The (CS2)2 dimer is a relatively simple molecular system composed of two carbon disulfide (CS2) molecules interacting with each other through intermolecular forces. Despite its apparent simplicity, the (CS2)2 dimer exhibits a rich array of structural and dynamical properties that are of great interest to researchers. In this research, we present an ab initio study of the intermolecular interactions in the carbon disulfide dimer (CS2)2 using an improved Lennard–Jones (ILJ) potential with CCSD(T)/QZVPP calculations. The potential energy surface of (CS2)2 is calculated using high-level quantum mechanical calculations based on the CCSD(T)/def2-qzvpp method, which accurately accounts for electron correlation effects. The resulting potential energy surface is then fitted to an ILJ potential energy function, which includes both long-range dipole–dipole interactions and short-range repulsive interactions. The calculated potential energy surface reveals a rich variety of structural and dynamical properties of (CS2)2, including multiple minima and saddle points, which are sensitive to the relative orientation of the two CS2 molecules. It is essential to use extended basis sets to accurately incorporate the significant quadrupole moment of CS2, which we have calculated to be 2.44 a.u. The results of this study demonstrate the importance of using high-level ab initio methods for the accurate calculation of potential energy surfaces in complex molecular systems such as (CS2)2. The use of an ILJ potential, which takes into account both dipole–dipole interactions and short-range repulsive interactions, provides a more accurate and efficient approach for modeling intermolecular interactions in (CS2)2 and other similar systems. The results of this study will be useful for understanding the behavior of carbon disulfide dimers in different environments and for the development of new materials and chemical processes.
期刊介绍:
The Journal of Physical Organic Chemistry is the foremost international journal devoted to the relationship between molecular structure and chemical reactivity in organic systems. It publishes Research Articles, Reviews and Mini Reviews based on research striving to understand the principles governing chemical structures in relation to activity and transformation with physical and mathematical rigor, using results derived from experimental and computational methods. Physical Organic Chemistry is a central and fundamental field with multiple applications in fields such as molecular recognition, supramolecular chemistry, catalysis, photochemistry, biological and material sciences, nanotechnology and surface science.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


