Catarina Falcão de Campos, Miguel Oliveira Santos, Rafael Roque, Isabel Conceição, Mamede de Carvalho
{"title":"线粒体神经胃肠道脑肌病:佛得角群岛患者胸苷磷酸化酶基因的新致病性突变。","authors":"Catarina Falcão de Campos, Miguel Oliveira Santos, Rafael Roque, Isabel Conceição, Mamede de Carvalho","doi":"10.1155/2019/5976410","DOIUrl":null,"url":null,"abstract":"<p><p>Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE) is a rare autosomal recessive disorder caused by mutations in the gene encoding the Thymidine Phosphorylase (TP). It is clinically characterized by severe gastrointestinal dysmotility, cachexia, palpebral ptosis, ophthalmoparesis, sensorimotor polyneuropathy and leukoencephalopathy. The diagnosis is established by the presence of typical clinical and neuroimaging features, positive family history, and abnormal genetic test. A 19-year-old Cape Verdean patient with a history since childhood of recurrent episodes of nausea, vomiting, diarrhoea and painful abdominal distension associated with progressive motor disability with difficulty in climbing stairs and running and clumsiness with her hands. The diagnostic workup was suggestive of MNGIE. Genetic screening of the <i>TYMP</i> gene identified a novel mutation (c. 1283 G>A). Patients with MNGIE have significant comorbidity and mortality, and they are frequently misdiagnosed. A better acknowledgment of this disorder is essential to permit an earlier diagnosis and to improve disease management.</p>","PeriodicalId":9615,"journal":{"name":"Case Reports in Neurological Medicine","volume":"2019 ","pages":"5976410"},"PeriodicalIF":0.9000,"publicationDate":"2019-12-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2019/5976410","citationCount":"6","resultStr":"{\"title\":\"Mitochondrial Neurogastrointestinal Encephalomyopathy: Novel Pathogenic Mutation in Thymidine Phosphorylase Gene in a Patient from Cape Verde Islands.\",\"authors\":\"Catarina Falcão de Campos, Miguel Oliveira Santos, Rafael Roque, Isabel Conceição, Mamede de Carvalho\",\"doi\":\"10.1155/2019/5976410\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE) is a rare autosomal recessive disorder caused by mutations in the gene encoding the Thymidine Phosphorylase (TP). It is clinically characterized by severe gastrointestinal dysmotility, cachexia, palpebral ptosis, ophthalmoparesis, sensorimotor polyneuropathy and leukoencephalopathy. The diagnosis is established by the presence of typical clinical and neuroimaging features, positive family history, and abnormal genetic test. A 19-year-old Cape Verdean patient with a history since childhood of recurrent episodes of nausea, vomiting, diarrhoea and painful abdominal distension associated with progressive motor disability with difficulty in climbing stairs and running and clumsiness with her hands. The diagnostic workup was suggestive of MNGIE. Genetic screening of the <i>TYMP</i> gene identified a novel mutation (c. 1283 G>A). Patients with MNGIE have significant comorbidity and mortality, and they are frequently misdiagnosed. A better acknowledgment of this disorder is essential to permit an earlier diagnosis and to improve disease management.</p>\",\"PeriodicalId\":9615,\"journal\":{\"name\":\"Case Reports in Neurological Medicine\",\"volume\":\"2019 \",\"pages\":\"5976410\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2019-12-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1155/2019/5976410\",\"citationCount\":\"6\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Neurological Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2019/5976410\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2019/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Neurological Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2019/5976410","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Mitochondrial Neurogastrointestinal Encephalomyopathy: Novel Pathogenic Mutation in Thymidine Phosphorylase Gene in a Patient from Cape Verde Islands.
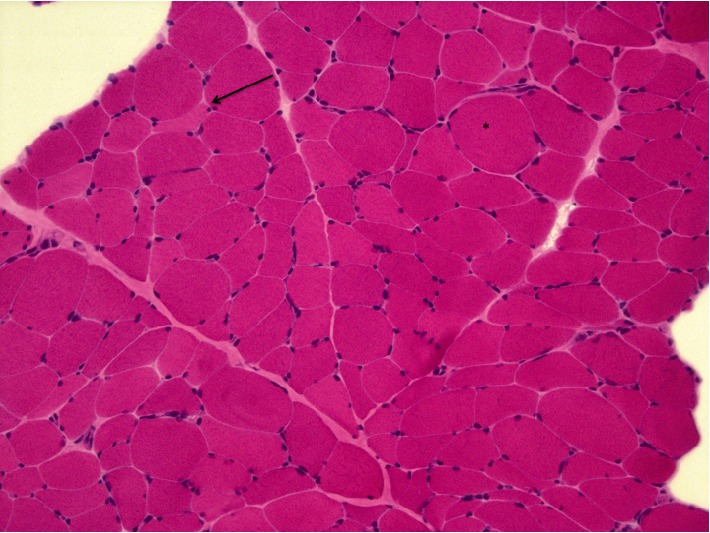
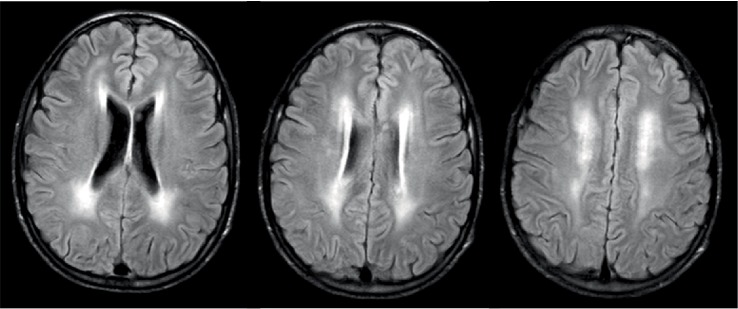
Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE) is a rare autosomal recessive disorder caused by mutations in the gene encoding the Thymidine Phosphorylase (TP). It is clinically characterized by severe gastrointestinal dysmotility, cachexia, palpebral ptosis, ophthalmoparesis, sensorimotor polyneuropathy and leukoencephalopathy. The diagnosis is established by the presence of typical clinical and neuroimaging features, positive family history, and abnormal genetic test. A 19-year-old Cape Verdean patient with a history since childhood of recurrent episodes of nausea, vomiting, diarrhoea and painful abdominal distension associated with progressive motor disability with difficulty in climbing stairs and running and clumsiness with her hands. The diagnostic workup was suggestive of MNGIE. Genetic screening of the TYMP gene identified a novel mutation (c. 1283 G>A). Patients with MNGIE have significant comorbidity and mortality, and they are frequently misdiagnosed. A better acknowledgment of this disorder is essential to permit an earlier diagnosis and to improve disease management.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


