Min-Sun Kim, Ari Song, Minji Im, June Huh, I-Seok Kang, Jinyoung Song, Aram Yang, Jinsup Kim, Eun-Kyung Kwon, Eu-Jin Choi, Sun-Ju Han, Hyung-Doo Park, Sung Yoon Cho, Dong-Kyu Jin
{"title":"韩国婴儿和晚发性庞贝病患儿的临床和分子特征:10年单中心酶替代治疗经验","authors":"Min-Sun Kim, Ari Song, Minji Im, June Huh, I-Seok Kang, Jinyoung Song, Aram Yang, Jinsup Kim, Eun-Kyung Kwon, Eu-Jin Choi, Sun-Ju Han, Hyung-Doo Park, Sung Yoon Cho, Dong-Kyu Jin","doi":"10.3345/kjp.2018.06968","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose: </strong>Pompe disease (PD) is an autosomal recessive disorder caused by a deficiency of acid alphaglucosidase resulting from pathogenic GAA variants. This study describes the clinical features, genotypes, changes before and after enzyme replacement therapy (ERT), and long-term outcomes in patients with infantile-onset PD (IOPD) and late-onset PD (LOPD) at a tertiary medical center.</p><p><strong>Methods: </strong>The medical records of 5 Korean patients (2 male, 3 female patients) diagnosed with PD between 2002 and 2013 at Samsung Medical Center in Seoul, Republic of Korea were retrospectively reviewed for data, including clinical and genetic characteristics at diagnosis and clinical course after ERT.</p><p><strong>Results: </strong>Common initial symptoms included hypotonia, cyanosis, and tachycardia in patients with IOPD and limb girdle weakness in patients with LOPD. Electrocardiography at diagnosis revealed hypertrophic cardiomyopathy in all patients with IOPD who showed a stable disease course during a median follow-up period of 10 years. Patients with LOPD showed improved hepatomegaly and liver transaminase level after ERT.</p><p><strong>Conclusion: </strong>As ERT is effective for treatment of PD, early identification of this disease is very important. Thus, patients with IOPD should be considered candidates for clinical trials of new drugs in the future.</p>","PeriodicalId":17863,"journal":{"name":"Korean Journal of Pediatrics","volume":"62 6","pages":"224-234"},"PeriodicalIF":0.0000,"publicationDate":"2019-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/85/f4/kjp-2018-06968.PMC6584236.pdf","citationCount":"6","resultStr":"{\"title\":\"Clinical and molecular characterization of Korean children with infantile and late-onset Pompe disease: 10 years of experience with enzyme replacement therapy at a single center.\",\"authors\":\"Min-Sun Kim, Ari Song, Minji Im, June Huh, I-Seok Kang, Jinyoung Song, Aram Yang, Jinsup Kim, Eun-Kyung Kwon, Eu-Jin Choi, Sun-Ju Han, Hyung-Doo Park, Sung Yoon Cho, Dong-Kyu Jin\",\"doi\":\"10.3345/kjp.2018.06968\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Purpose: </strong>Pompe disease (PD) is an autosomal recessive disorder caused by a deficiency of acid alphaglucosidase resulting from pathogenic GAA variants. This study describes the clinical features, genotypes, changes before and after enzyme replacement therapy (ERT), and long-term outcomes in patients with infantile-onset PD (IOPD) and late-onset PD (LOPD) at a tertiary medical center.</p><p><strong>Methods: </strong>The medical records of 5 Korean patients (2 male, 3 female patients) diagnosed with PD between 2002 and 2013 at Samsung Medical Center in Seoul, Republic of Korea were retrospectively reviewed for data, including clinical and genetic characteristics at diagnosis and clinical course after ERT.</p><p><strong>Results: </strong>Common initial symptoms included hypotonia, cyanosis, and tachycardia in patients with IOPD and limb girdle weakness in patients with LOPD. Electrocardiography at diagnosis revealed hypertrophic cardiomyopathy in all patients with IOPD who showed a stable disease course during a median follow-up period of 10 years. Patients with LOPD showed improved hepatomegaly and liver transaminase level after ERT.</p><p><strong>Conclusion: </strong>As ERT is effective for treatment of PD, early identification of this disease is very important. Thus, patients with IOPD should be considered candidates for clinical trials of new drugs in the future.</p>\",\"PeriodicalId\":17863,\"journal\":{\"name\":\"Korean Journal of Pediatrics\",\"volume\":\"62 6\",\"pages\":\"224-234\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2019-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/85/f4/kjp-2018-06968.PMC6584236.pdf\",\"citationCount\":\"6\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Korean Journal of Pediatrics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3345/kjp.2018.06968\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2018/10/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Korean Journal of Pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3345/kjp.2018.06968","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/10/4 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Clinical and molecular characterization of Korean children with infantile and late-onset Pompe disease: 10 years of experience with enzyme replacement therapy at a single center.
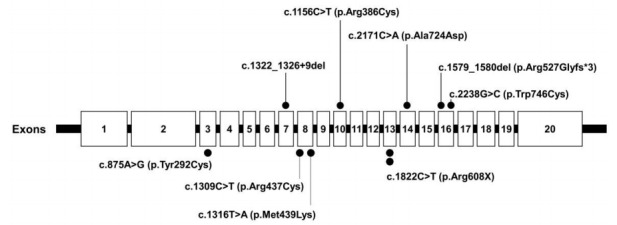
Purpose: Pompe disease (PD) is an autosomal recessive disorder caused by a deficiency of acid alphaglucosidase resulting from pathogenic GAA variants. This study describes the clinical features, genotypes, changes before and after enzyme replacement therapy (ERT), and long-term outcomes in patients with infantile-onset PD (IOPD) and late-onset PD (LOPD) at a tertiary medical center.
Methods: The medical records of 5 Korean patients (2 male, 3 female patients) diagnosed with PD between 2002 and 2013 at Samsung Medical Center in Seoul, Republic of Korea were retrospectively reviewed for data, including clinical and genetic characteristics at diagnosis and clinical course after ERT.
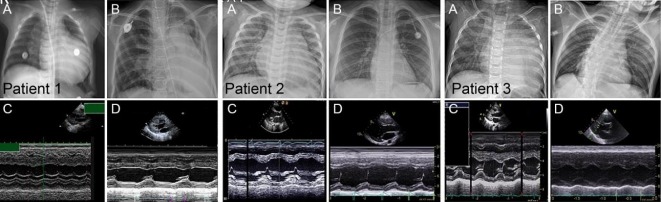
Results: Common initial symptoms included hypotonia, cyanosis, and tachycardia in patients with IOPD and limb girdle weakness in patients with LOPD. Electrocardiography at diagnosis revealed hypertrophic cardiomyopathy in all patients with IOPD who showed a stable disease course during a median follow-up period of 10 years. Patients with LOPD showed improved hepatomegaly and liver transaminase level after ERT.
Conclusion: As ERT is effective for treatment of PD, early identification of this disease is very important. Thus, patients with IOPD should be considered candidates for clinical trials of new drugs in the future.
期刊介绍:
Korean J Pediatr covers clinical and research works relevant to all aspects of child healthcare. The journal aims to serve pediatricians through the prompt publication of significant advances in any field of pediatrics and to rapidly disseminate recently updated knowledge to the public. Additionally, it will initiate dynamic, international, academic discussions concerning the major topics related to pediatrics. Manuscripts are categorized as review articles, original articles, and case reports. Areas of specific interest include: Growth and development, Neonatology, Pediatric neurology, Pediatric nephrology, Pediatric endocrinology, Pediatric cardiology, Pediatric allergy, Pediatric pulmonology, Pediatric infectious diseases, Pediatric immunology, Pediatric hemato-oncology, Pediatric gastroenterology, Nutrition, Human genetics, Metabolic diseases, Adolescence medicine, General pediatrics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


