{"title":"从遗传学和表观遗传学角度看多发性内分泌肿瘤综合征。","authors":"Fatemeh Khatami, Seyed Mohammad Tavangar","doi":"10.1177/1177271918785129","DOIUrl":null,"url":null,"abstract":"<p><p>Multiple endocrine neoplasia (MEN) syndromes are infrequent inherited disorders in which more than one endocrine glands develop noncancerous (benign) or cancerous (malignant) tumors or grow excessively without forming tumors. There are 3 famous and well-known forms of MEN syndromes (MEN 1, MEN 2A, and MEN 2B) and a newly documented one (MEN4). These syndromes are infrequent and occurred in all ages and both men and women. Usually, germ line mutations that can be resulted in neoplastic transformation of anterior pituitary, parathyroid glands, and pancreatic islets in addition to gastrointestinal tract can be an indicator for MEN1. The medullary thyroid cancer (MTC) in association with pheochromocytoma and/or multiple lesions of parathyroid glands with hyperparathyroidism can be pointer of MEN2 which can be subgrouped into the MEN 2A, MEN 2B, and familial MTC syndromes. There are no distinct biochemical markers that allow identification of familial versus nonfamilial forms of the tumors, but familial MTC usually happens at a younger age than sporadic MTC. The <i>MEN1</i> gene (menin protein) is in charge of MEN 1 disease, CDNK1B for MEN 4, and RET proto-oncogene for MEN 2. The focus over the molecular targets can bring some hope for both diagnosis and management of MEN syndromes. In the current review, we look at this disease and responsible genes and their cell signaling pathway involved.</p>","PeriodicalId":47060,"journal":{"name":"Biomarker Insights","volume":"13 ","pages":"1177271918785129"},"PeriodicalIF":3.4000,"publicationDate":"2018-07-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/7b/c5/10.1177_1177271918785129.PMC6043927.pdf","citationCount":"0","resultStr":"{\"title\":\"Multiple Endocrine Neoplasia Syndromes from Genetic and Epigenetic Perspectives.\",\"authors\":\"Fatemeh Khatami, Seyed Mohammad Tavangar\",\"doi\":\"10.1177/1177271918785129\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Multiple endocrine neoplasia (MEN) syndromes are infrequent inherited disorders in which more than one endocrine glands develop noncancerous (benign) or cancerous (malignant) tumors or grow excessively without forming tumors. There are 3 famous and well-known forms of MEN syndromes (MEN 1, MEN 2A, and MEN 2B) and a newly documented one (MEN4). These syndromes are infrequent and occurred in all ages and both men and women. Usually, germ line mutations that can be resulted in neoplastic transformation of anterior pituitary, parathyroid glands, and pancreatic islets in addition to gastrointestinal tract can be an indicator for MEN1. The medullary thyroid cancer (MTC) in association with pheochromocytoma and/or multiple lesions of parathyroid glands with hyperparathyroidism can be pointer of MEN2 which can be subgrouped into the MEN 2A, MEN 2B, and familial MTC syndromes. There are no distinct biochemical markers that allow identification of familial versus nonfamilial forms of the tumors, but familial MTC usually happens at a younger age than sporadic MTC. The <i>MEN1</i> gene (menin protein) is in charge of MEN 1 disease, CDNK1B for MEN 4, and RET proto-oncogene for MEN 2. The focus over the molecular targets can bring some hope for both diagnosis and management of MEN syndromes. In the current review, we look at this disease and responsible genes and their cell signaling pathway involved.</p>\",\"PeriodicalId\":47060,\"journal\":{\"name\":\"Biomarker Insights\",\"volume\":\"13 \",\"pages\":\"1177271918785129\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2018-07-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/7b/c5/10.1177_1177271918785129.PMC6043927.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biomarker Insights\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/1177271918785129\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2018/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biomarker Insights","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/1177271918785129","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
摘要
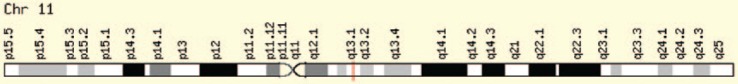
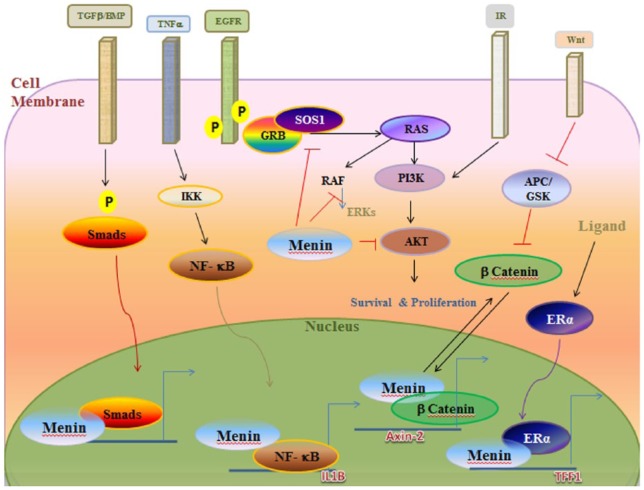
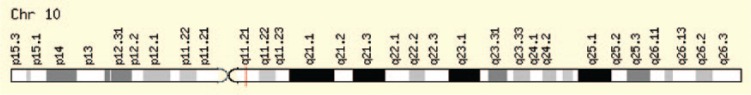
多发性内分泌肿瘤(MEN)综合征是一种不常见的遗传性疾病,患者的一个以上内分泌腺会出现非癌(良性)或癌(恶性)肿瘤,或过度生长而不形成肿瘤。有三种著名的 MEN 综合征(MEN 1、MEN 2A 和 MEN 2B)和一种新发现的 MEN 综合征(MEN4)。这些综合征并不常见,男女老幼均可发病。通常,可导致垂体前叶、甲状旁腺、胰岛以及胃肠道肿瘤性转化的种系突变可作为 MEN1 的指标。甲状腺髓样癌与嗜铬细胞瘤和/或甲状旁腺多发性病变合并甲状旁腺功能亢进可作为MEN2的指针,而MEN2又可细分为MEN2A、MEN2B和家族性甲状腺髓样癌综合征。目前还没有明显的生化标志物可用于鉴别家族性与非家族性肿瘤,但家族性 MTC 的发病年龄通常比散发性 MTC 要小。MEN1 基因(menin 蛋白)负责 MEN1 疾病,CDNK1B 负责 MEN4,RET 原癌基因负责 MEN2。对分子靶点的关注为 MEN 综合征的诊断和治疗带来了希望。在本综述中,我们将探讨这种疾病及其相关基因和细胞信号通路。
Multiple Endocrine Neoplasia Syndromes from Genetic and Epigenetic Perspectives.
Multiple endocrine neoplasia (MEN) syndromes are infrequent inherited disorders in which more than one endocrine glands develop noncancerous (benign) or cancerous (malignant) tumors or grow excessively without forming tumors. There are 3 famous and well-known forms of MEN syndromes (MEN 1, MEN 2A, and MEN 2B) and a newly documented one (MEN4). These syndromes are infrequent and occurred in all ages and both men and women. Usually, germ line mutations that can be resulted in neoplastic transformation of anterior pituitary, parathyroid glands, and pancreatic islets in addition to gastrointestinal tract can be an indicator for MEN1. The medullary thyroid cancer (MTC) in association with pheochromocytoma and/or multiple lesions of parathyroid glands with hyperparathyroidism can be pointer of MEN2 which can be subgrouped into the MEN 2A, MEN 2B, and familial MTC syndromes. There are no distinct biochemical markers that allow identification of familial versus nonfamilial forms of the tumors, but familial MTC usually happens at a younger age than sporadic MTC. The MEN1 gene (menin protein) is in charge of MEN 1 disease, CDNK1B for MEN 4, and RET proto-oncogene for MEN 2. The focus over the molecular targets can bring some hope for both diagnosis and management of MEN syndromes. In the current review, we look at this disease and responsible genes and their cell signaling pathway involved.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


