{"title":"针对沃尔曼病和胆固醇酯沉积病:疾病发病机制和治疗进展。","authors":"Francis Aguisanda, Natasha Thorne, Wei Zheng","doi":"10.2174/2213988501711010001","DOIUrl":null,"url":null,"abstract":"<p><p>Wolman disease (WD) and cholesteryl ester storage disease (CESD) are lysosomal storage diseases (LSDs) caused by a deficiency in lysosomal acid lipase (LAL) due to mutations in the <i>LIPA</i> gene. This enzyme is critical to the proper degradation of cholesterol in the lysosome. LAL function is completely lost in WD while some residual activity remains in CESD. Both are rare diseases with an incidence rate of less than 1/100,000 births for WD and approximate 2.5/100,000 births for CESD. Clinical manifestation of WD includes hepatosplenomegaly, calcified adrenal glands, severe malabsorption and a failure to thrive. As in CESD, histological analysis of WD tissues reveals the accumulation of triglycerides (TGs) and esterified cholesterol (EC) in cellular lysosomes. However, the clinical presentation of CESD is less severe and more variable than WD. This review is to provide an overview of the disease pathophysiology and the current state of therapeutic development for both of WD and CESD. The review will also discuss the application of patient derived iPSCs for further drug discovery.</p>","PeriodicalId":10755,"journal":{"name":"Current Chemical Genomics and Translational Medicine","volume":"11 ","pages":"1-18"},"PeriodicalIF":0.0000,"publicationDate":"2017-01-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.2174/2213988501711010001","citationCount":"39","resultStr":"{\"title\":\"Targeting Wolman Disease and Cholesteryl Ester Storage Disease: Disease Pathogenesis and Therapeutic Development.\",\"authors\":\"Francis Aguisanda, Natasha Thorne, Wei Zheng\",\"doi\":\"10.2174/2213988501711010001\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Wolman disease (WD) and cholesteryl ester storage disease (CESD) are lysosomal storage diseases (LSDs) caused by a deficiency in lysosomal acid lipase (LAL) due to mutations in the <i>LIPA</i> gene. This enzyme is critical to the proper degradation of cholesterol in the lysosome. LAL function is completely lost in WD while some residual activity remains in CESD. Both are rare diseases with an incidence rate of less than 1/100,000 births for WD and approximate 2.5/100,000 births for CESD. Clinical manifestation of WD includes hepatosplenomegaly, calcified adrenal glands, severe malabsorption and a failure to thrive. As in CESD, histological analysis of WD tissues reveals the accumulation of triglycerides (TGs) and esterified cholesterol (EC) in cellular lysosomes. However, the clinical presentation of CESD is less severe and more variable than WD. This review is to provide an overview of the disease pathophysiology and the current state of therapeutic development for both of WD and CESD. The review will also discuss the application of patient derived iPSCs for further drug discovery.</p>\",\"PeriodicalId\":10755,\"journal\":{\"name\":\"Current Chemical Genomics and Translational Medicine\",\"volume\":\"11 \",\"pages\":\"1-18\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2017-01-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.2174/2213988501711010001\",\"citationCount\":\"39\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current Chemical Genomics and Translational Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2174/2213988501711010001\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2017/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Chemical Genomics and Translational Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2174/2213988501711010001","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2017/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Targeting Wolman Disease and Cholesteryl Ester Storage Disease: Disease Pathogenesis and Therapeutic Development.
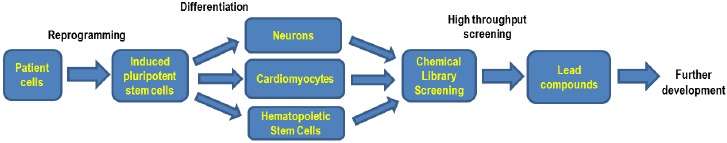
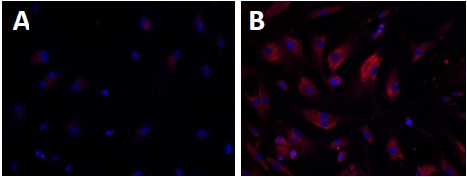
Wolman disease (WD) and cholesteryl ester storage disease (CESD) are lysosomal storage diseases (LSDs) caused by a deficiency in lysosomal acid lipase (LAL) due to mutations in the LIPA gene. This enzyme is critical to the proper degradation of cholesterol in the lysosome. LAL function is completely lost in WD while some residual activity remains in CESD. Both are rare diseases with an incidence rate of less than 1/100,000 births for WD and approximate 2.5/100,000 births for CESD. Clinical manifestation of WD includes hepatosplenomegaly, calcified adrenal glands, severe malabsorption and a failure to thrive. As in CESD, histological analysis of WD tissues reveals the accumulation of triglycerides (TGs) and esterified cholesterol (EC) in cellular lysosomes. However, the clinical presentation of CESD is less severe and more variable than WD. This review is to provide an overview of the disease pathophysiology and the current state of therapeutic development for both of WD and CESD. The review will also discuss the application of patient derived iPSCs for further drug discovery.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


