{"title":"基于反应坐标的速率理论对化学反应建模的准确性:来自视网膜热异构化的见解","authors":"Simon Ghysbrecht, Luca Donati, Bettina G. Keller","doi":"10.1002/jcc.27529","DOIUrl":null,"url":null,"abstract":"<p>Modern potential energy surfaces have shifted attention to molecular simulations of chemical reactions. While various methods can estimate rate constants for conformational transitions in molecular dynamics simulations, their applicability to studying chemical reactions remains uncertain due to the high and sharp energy barriers and complex reaction coordinates involved. This study focuses on the thermal cis-trans isomerization in retinal, employing molecular simulations and comparing rate constant estimates based on one-dimensional rate theories with those based on sampling transitions and grid-based models for low-dimensional collective variable spaces. Even though each individual method to estimate the rate passes its quality tests, the rate constant estimates exhibit considerable disparities. Rate constant estimates based on one-dimensional reaction coordinates prove challenging to converge, even if the reaction coordinate is optimized. However, consistent estimates of the rate constant are achieved by sampling transitions and by multi-dimensional grid-based models.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-12-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27529","citationCount":"0","resultStr":"{\"title\":\"Accuracy of Reaction Coordinate Based Rate Theories for Modelling Chemical Reactions: Insights From the Thermal Isomerization in Retinal\",\"authors\":\"Simon Ghysbrecht, Luca Donati, Bettina G. Keller\",\"doi\":\"10.1002/jcc.27529\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Modern potential energy surfaces have shifted attention to molecular simulations of chemical reactions. While various methods can estimate rate constants for conformational transitions in molecular dynamics simulations, their applicability to studying chemical reactions remains uncertain due to the high and sharp energy barriers and complex reaction coordinates involved. This study focuses on the thermal cis-trans isomerization in retinal, employing molecular simulations and comparing rate constant estimates based on one-dimensional rate theories with those based on sampling transitions and grid-based models for low-dimensional collective variable spaces. Even though each individual method to estimate the rate passes its quality tests, the rate constant estimates exhibit considerable disparities. Rate constant estimates based on one-dimensional reaction coordinates prove challenging to converge, even if the reaction coordinate is optimized. However, consistent estimates of the rate constant are achieved by sampling transitions and by multi-dimensional grid-based models.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"46 1\",\"pages\":\"\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-12-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27529\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27529\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27529","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Accuracy of Reaction Coordinate Based Rate Theories for Modelling Chemical Reactions: Insights From the Thermal Isomerization in Retinal
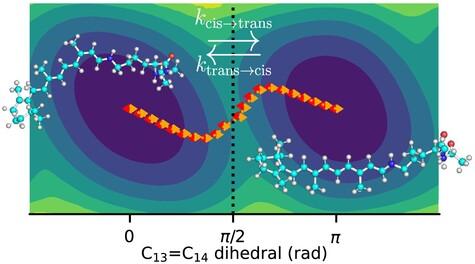
Modern potential energy surfaces have shifted attention to molecular simulations of chemical reactions. While various methods can estimate rate constants for conformational transitions in molecular dynamics simulations, their applicability to studying chemical reactions remains uncertain due to the high and sharp energy barriers and complex reaction coordinates involved. This study focuses on the thermal cis-trans isomerization in retinal, employing molecular simulations and comparing rate constant estimates based on one-dimensional rate theories with those based on sampling transitions and grid-based models for low-dimensional collective variable spaces. Even though each individual method to estimate the rate passes its quality tests, the rate constant estimates exhibit considerable disparities. Rate constant estimates based on one-dimensional reaction coordinates prove challenging to converge, even if the reaction coordinate is optimized. However, consistent estimates of the rate constant are achieved by sampling transitions and by multi-dimensional grid-based models.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


