从 GSK2256098 类似物中建立 FAK-PROTAC 候选者模型,用于靶向蛋白质降解。
IF 2.5
3区 生物学
Q3 BIOCHEMISTRY & MOLECULAR BIOLOGY
Biochemical and biophysical research communications
Pub Date : 2024-11-17
DOI:10.1016/j.bbrc.2024.151001
引用次数: 0
摘要
通过传统的药物设计方法抑制蛋白质已被证明是开发众多基于小分子疗法的有效方法。近十年来,小分子抑制剂引导的蛋白质降解作为一种替代方法应运而生,有望满足无法药物靶点的药物需求。病灶粘附激酶(FAK)是肿瘤生长和扩散的关键调节因子,此外它还是其他蛋白质信号传导的支架。迄今为止,FAK 抑制剂在癌症临床试验中的应用效果并不理想。与之前控制 FAK 表达的尝试不同的是,之前的尝试仅限于激酶域抑制,在临床研究中取得的成功有限,而蛋白降解则有可能同时破坏 FAK 的激酶和支架功能。最近,一些基于 FAK I 型抑制剂、采用复杂化学合成方法的 FAK 降解剂被报道。有趣的是,最近发表的一种三元复合物揭示了 FAK-PROTAC-E3 复合物的结合模式。该复合物为开发针对 FAK 蛋白的合理 PROTAC 设计开辟了一条途径。因此,在本研究中,我们选择了活性最强的 I 型 FAK 抑制剂 GSK2256098。该抑制剂的结合模式促使我们为 PROTAC 设计寻找最合适的类似物。通过应用分子对接(MD)和分子动力学模拟(MDS),我们确定了一种适合用于 PTOTAC 设计的高亲和力类似物。此外,在 FAK-PROTAC-E3 三元复合物的基础上,我们还在这两种蛋白之间构建了二元复合物 FAK-Hit-E3-VHL。利用基于结构的方法,我们设计了十种不同的潜在 FAK PROTACs 候选物。使用 MDS 分析了复合物的稳定性,并利用结合自由能预测了结合亲和力。最后,与已知的 FAK PROTAC GSK215 相比,基于与目标蛋白和 E3 连接酶之间理想的分子间相互作用,ProTAC4 被选为最佳候选。本文章由计算机程序翻译,如有差异,请以英文原文为准。
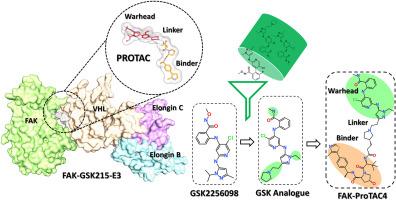
Modeling of FAK-PROTAC candidates from GSK2256098 analogs for targeted protein degradation
Protein inhibition via the traditional drug-designing approach has been shown to be an effective method for developing numerous small-molecule-based therapeutics. In the last decade, small inhibitors-guided protein degradation has arisen as an alternative method with the potential to fulfill the drug requirement for undruggable targets. Focal adhesion kinase (FAK) is a crucial modulator of the growth and spread of tumors, apart from it also acts as a scaffold for signaling of other proteins. FAK inhibitors have thus far had unsatisfactory results in clinical trials for cancer applications. Unlike prior attempts to control FAK expression, which were restricted to kinase domain inhibition with limited success in clinical research, protein degradation has the potential to concurrently disrupt FAK's kinase and scaffolding function. Recently, several FAK degraders were reported based on FAK Type I inhibitors using complex chemical synthesis approaches. Interestingly, recently a ternary complex was published revealing the binding mode of the FAK-PROTAC-E3 complex. This complex opens an avenue for the development of rational PROTAC design against FAK protein. Therefore, in the present study, we selected the most active Type I FAK inhibitor GSK2256098. The binding mode of the inhibitor prompted us to identify the most suitable analog for PROTAC design. We have identified a high-affinity analog that is suitable for PTOTAC design through the application of molecular docking (MD) and molecular dynamics simulations (MDS). Further based on the ternary FAK-PROTAC-E3 complex we build a binary complex FAK-Hit-E3-VHL between both proteins. Using the structure-based approach ten different potential FAK PROTACs candidates were designed. The stability of the complexes was analyzed using MDS and binding free energies were used to predict the binding affinity. Finally, based on desirable intermolecular interactions with the target and E3 ligase ProTAC4 was selected as the best candidate when compared with known FAK PROTAC GSK215.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
6.10
自引率
0.00%
发文量
1400
审稿时长
14 days
期刊介绍:
Biochemical and Biophysical Research Communications is the premier international journal devoted to the very rapid dissemination of timely and significant experimental results in diverse fields of biological research. The development of the "Breakthroughs and Views" section brings the minireview format to the journal, and issues often contain collections of special interest manuscripts. BBRC is published weekly (52 issues/year).Research Areas now include: Biochemistry; biophysics; cell biology; developmental biology; immunology
; molecular biology; neurobiology; plant biology and proteomics
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


