Paola Coppola, Eva Gil Berglund, Karen Rowland Yeo
{"title":"孕期用药:临床药理学外推框架,弥补知识空白。","authors":"Paola Coppola, Eva Gil Berglund, Karen Rowland Yeo","doi":"10.1002/psp4.13242","DOIUrl":null,"url":null,"abstract":"<p>Drug treatment may be required during pregnancy, both for pregnant women and their unborn children. About 6 million pregnancies in the United States (US) occur each year, with most women taking at least one prescription medication during pregnancy and more than half of the mothers taking medicines after delivery (Pregnant?_Breastfeeding?_FDA_Aims_to_Improve_Drug_Information_[fda.gov]). However, in our attempts to protect the unborn children or breastfeeding infants, information to support such treatment is rarely generated and drugs are often used off-label.</p><p>Systematic exclusion of pregnant women from clinical trials at all stages does not allow the collection of data to support the safe use of medicines during pregnancy. Dosing strategies to treat health conditions developed either before or during pregnancy often rely on data from healthy and/or nonpregnant subjects, instead of being driven by complex pregnancy-related physiological changes on drug exposure. Despite the recognized medical need, a recent review captured labels with clinically meaningful interventions in pregnancy for only 139 medications in the US and 20 in the European Union (EU); in both cases, 30%–40% had established doses for pregnant populations.<span><sup>1</sup></span> Information on dosing during pregnancy is often unavailable in original regulatory submissions limiting label recommendation. Pre-authorization data in pregnant population is generally not requested and post-authorization registry studies are mainly required for drugs where substantial use during pregnancy is foreseen, for example, in malaria or for HIV treatment, monitoring pregnancy outcomes in women exposed to drugs during gestation (Postmarket_Requirements_and_Commitments_[fda.gov]). In addition to data collection in registries, when pregnancy is expected to impact systemic drug levels, clinical PK data are generated post-authorization to inform dosing recommendations (e.g., rilpivirine, darunavir, cobicistat).</p><p>For years, the clinical need for drug treatment during pregnancy has been left largely unresolved by regulators and sponsors, leaving the risk–benefit assessment to prescribers and patients. However, health care and community attention to this unmet medical need has resulted in increased regulatory action. In 2018, the FDA published a draft guidance on Scientific and Ethical Considerations for the Inclusion of pregnant women in Clinical Trials. In 2022, current thinking and regulatory efforts were communicated by Sewell et al.<span><sup>2</sup></span> and the FDA diversity plan framework (Diversity_Plans_to_Improve_Enrollment_of_Participants_from_Underrepresented_Racial_and_Ethnic_Populations_in_Clinical_Trials_Guidance_for_Industry_(fda.gov)). Furthermore, regulators from the FDA, EMA, and MHRA acknowledged the urgent need to shift from systematic exclusion to the inclusion of pregnant and breastfeeding women in clinical trials at the International Coalition of Medicines Regulatory Authorities Pregnancy and Lactation Workshop (ICMRA_Pregnancy_and_Lactation_Workshop_International_Coalition_of_Medicines_Regulatory_Authorities_(ICMRA)). It was proposed that applicants should develop and submit a “Maternal Investigation Plan,” outlining the strategy to study these populations. This change of approach requires international collaboration and harmonization, and foundations were laid for the development of the ICH21 Guideline (ICH_E21_Final_Concept_Paper_2023) which will outline the investigational development plan, alongside other factors considered in the extrapolation framework of the draft ICH E11 guideline for pediatrics (draft-ich-guideline-e11a-pediatric-extrapolation-step-2b_en.pdf_(europa.eu)). The FDA has also held workshops to encourage an increase in the number of studies conducted in pregnant woman (Pharmacokinetic_Evaluation_in_Pregnancy_(fda.gov); Fetal_Pharmacology_and_Therapeutics_(fda.gov)).</p><p>Pregnancy-related physiological changes can affect drug PK resulting in possible loss of efficacy, or potential toxicity in both the mother and fetus. Phase I (e.g., CYP3A4, CYP2D6, and CYP219) and phase II (UGT1A1 and UGT1A4) metabolizing enzyme activities are altered during pregnancy. This is illustrated by the observed ~60% increase in clearance and ~60% decrease in exposure of the CYP2D6 substrates metoprolol and fluoxetine, respectively.<span><sup>3, 4</sup></span> Corresponding decreases in exposure have been reported for the CYP3A substrates rilpivirine and cobicistat-boosted darunavir as well as for the UGT1A1 substrate dolutegravir. Increased glomerular filtration rate during pregnancy, can lead to reduced exposure of drugs undergoing renal excretion (e.g., ceftazidime).</p><p>In some cases, the decrease in exposure has been reflected in the labeling, driving either dose recommendations, monitoring, or contraindications.</p><p>In the original rilpivirine label (2011), the use of the drug during pregnancy was restricted to cases where the potential benefit justifies the potential risk. In 2018, the labeling was updated based on new data (i.e., 30%–40% lower exposure in second and third trimesters than postpartum), resulting in a recommendation to closely monitor viral load due to lower exposures during pregnancy (EDURANT®_(rilpivirine)_US_label).</p><p>Similarly, the exposure of dolutegravir was reported to be up to 37% lower during pregnancy.<span><sup>5</sup></span> In the recently updated label (2024), clinical fetal outcome data from observational studies supported the removal of pregnancy testing before initiation of treatment as well as the warning of embryo–fetal toxicity (TIVICAY_(dolutegravir)_US_label).</p><p>As an example of major labeling impact, the exposure of darunavir boosted with cobicistat was substantially lower (>50% decrease in AUC, and ~90% decrease in <i>C</i><sub>min</sub>) during the second and third trimesters. Hence, the label states that use during pregnancy is not recommended. An exposure registry monitors pregnancy outcomes in individuals taking the drugs (PREZCOBIX®_(darunavir_and_cobicistat)_US_label). Publicly available data are not sufficient to allow the evaluation of whether a dose increase may be feasible and safe for the mother and fetus.</p><p>The EMA recommends therapeutic drug monitoring for ritonavir-boosted atazanavir (300/100 mg) as insufficient exposure to atazanavir may occur in pregnant patients. Moreover, both EMA and FDA labels state that if either tenofovir or an H2-receptor antagonist is co-administered in pregnant women, an atazanavir dose increase (400 mg) may be required due to enhanced exposure reduction (REYATAZ_[atazanavir]_US_label; REYATAZ_EMA_SmPC).</p><p>Before these labeling changes were implemented, it is likely that uninformed, off-label use occurred.</p><p>The pediatric extrapolation framework outlined in ICH E11 (ICH_Guideline_E11A_on_Pediatric_Extrapolation) can be adapted to pregnancy, supporting decisions on the need for studies, informing study design and labeling update, and generating new data to fill the knowledge gaps. Particular consideration should be made for teratogenicity and fetal toxicity, and a cautious, informed, approach is recommended. Evaluation and interpretation of fetal and preclinical data informing risk should be considered but it is outside the scope of this paper. Registries provide important follow-up information on fetal, that is, pediatric safety detecting signals and allow broader scientific analysis. If the use of a medicine in pregnant women, during certain trimesters, is too rare for conventional studies to be possible, adapted strategies to obtain PK and clinical information should be planned for. Labeling language could be considered to enable these approaches.</p><p>As in ICH E11, the drug development plan should consider the clinical need for treatment and foreseen risks in each trimester as well as significant benefits compared with available therapies. The efficacy extrapolation approach could be based on similarity in disease and in response to treatment, drug pharmacology, expected similarity in target exposure, and how to reach this exposure in pregnant individuals. Scientific literature, preclinical and clinical data, clinical experience with other drugs, including RWE, and foreseen PK differences, should all be considered (Figure 1).</p><p>The extrapolation framework could support decisions on mother and fetal safety management including study design, sample size (which might be smaller than conventional phase III), generation of post-authorization safety data in a larger population, need for long-term follow-up and registries, etc. Key considerations also include risks to mother and fetus if not treating the disease, the need for gradual recruitment into earlier trimesters, available safety information in nonpregnant women, whether target/off-target effects could be different, or particularly impact pregnant women or the fetus. Safety data available for drugs with similar mechanisms of action, or off-target effects are crucial, as well as preclinical fetal toxicity/teratogenicity information and the need to follow-up specific short- and long-term effects, etc. (Table 1). Expected fetal exposure to drugs should if possible be considered.</p><p>Availability of data assessing similarity of exposure–response (ER) relationships for efficacy and safety compared with nonpregnant should be reflected in the study design, and physiological factors impacting drug exposure during pregnancy considered in dose selection, and significant uncertainties resolved by confirmatory “PK lead-in” investigations.</p><p>Sparse sampling approaches should be optimized using population PK (pop-PK) to support study designs, for example, informing sample size estimations. The use of physiologically-based pharmacokinetic (PBPK) modeling to simulate drug exposure is recommended for initial dose selection, as it allows multifactorial mechanistic knowledge to inform exposure predictions. For marketed drugs, the analysis of pharmacovigilance and real-world data on use during pregnancy should be considered. It is also important to understand whether loss of efficacy is expected due to altered drug systemic exposure, and if temporary dose adjustment is needed.</p><p>The clinical pharmacology strategy for investigating the impact of pregnancy on drug exposure requires integration of available data, including clinical PK data collected in nonpregnant populations, preclinical information, a good understanding of the effect of pregnancy-related physiological changes on drug exposure, potential DDIs, and exposure–response relationship. The totality of evidence can inform the development of a MIDD-based extrapolation framework to identify and fill knowledge gaps underlining a development strategy to optimize the use of available data.</p><p>The use of PBPK models, incorporating relevant physiological changes, to predict drug exposure and PD response (target and off-target pharmacological effects) in pregnant women and the fetus is being considered by regulators as a tool to possibly inform dosing during pregnancy and support benefit–risk decisions [ICH_E21_Final_Concept_Paper_2023].<span><sup>6, 7</sup></span></p><p>When there is a good understanding of the effect of pregnancy on the physiological factors impacting the disposition of the specific drug, PBPK modeling can be confidently applied to predict exposure changes and drive dosing decisions. Several authors showed that PBPK modeling can reliably predict those changes in exposure in pregnancy compared with nonpregnant populations for a range of drugs,<span><sup>8, 9</sup></span> [Pregnancy_Physiologically_Based_Pharmacokinetic_(PBPK)_Modeling_with_Population_Variability_for_Drug_Safety_and_Efficacy_Assessment_(fda.gov)]. A high-fidelity drug model<span><sup>10</sup></span> (where the elimination routes are clearly elucidated) developed initially in the nonpregnant population can then be applied to predict an initial dose in pregnant subjects. Once PK data in the pregnant patients have been accrued, pop-PK modeling could be used to support the PBPK modeling, quantify ER relationships, and analyze covariates affecting exposure.</p><p>A change of mindset appears to have been adopted by the scientific and regulatory community. The need for pregnant women to have well-informed medical treatments has been recognized, going from a protection from research to a protection by research strategy. The risk of fetal toxicity should be reflected in relevant parts of the development and multifactorial risk–benefit discussions. The regulatory framework could be based on approaches like global pediatric regulations. Indeed, we propose to adapt the pediatric development extrapolation framework, (ICH_Guideline_E11A_on_Pediatric_Extrapolation), providing a systematic approach to drug development, identifying important knowledge gaps to be addressed in the clinical study design and follow-up strategies. A clinical pharmacology strategy based on MIDD approaches, such as PBPK and pop-PK is central to this approach.</p><p>No funding was received for this work.</p><p>P.C. is an employee of Certara Italy (Certara Drug Development Solutions). E.B. is an employee of Certara NL (Certara Drug Development Solutions). K.R.Y. is an employee of Certara UK Limited (Simcyp Division).</p><p>As Deputy-Editor-Chief for <i>CPT: Pharmacometrics & Systems Pharmacology</i>, Karen Rowland Yeo was not involved in the review or decision-making processes for this work.</p>","PeriodicalId":10774,"journal":{"name":"CPT: Pharmacometrics & Systems Pharmacology","volume":"13 11","pages":"1830-1834"},"PeriodicalIF":3.1000,"publicationDate":"2024-10-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/psp4.13242","citationCount":"0","resultStr":"{\"title\":\"Medicines in pregnancy: A clinical pharmacology extrapolation framework to address knowledge gaps\",\"authors\":\"Paola Coppola, Eva Gil Berglund, Karen Rowland Yeo\",\"doi\":\"10.1002/psp4.13242\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Drug treatment may be required during pregnancy, both for pregnant women and their unborn children. About 6 million pregnancies in the United States (US) occur each year, with most women taking at least one prescription medication during pregnancy and more than half of the mothers taking medicines after delivery (Pregnant?_Breastfeeding?_FDA_Aims_to_Improve_Drug_Information_[fda.gov]). However, in our attempts to protect the unborn children or breastfeeding infants, information to support such treatment is rarely generated and drugs are often used off-label.</p><p>Systematic exclusion of pregnant women from clinical trials at all stages does not allow the collection of data to support the safe use of medicines during pregnancy. Dosing strategies to treat health conditions developed either before or during pregnancy often rely on data from healthy and/or nonpregnant subjects, instead of being driven by complex pregnancy-related physiological changes on drug exposure. Despite the recognized medical need, a recent review captured labels with clinically meaningful interventions in pregnancy for only 139 medications in the US and 20 in the European Union (EU); in both cases, 30%–40% had established doses for pregnant populations.<span><sup>1</sup></span> Information on dosing during pregnancy is often unavailable in original regulatory submissions limiting label recommendation. Pre-authorization data in pregnant population is generally not requested and post-authorization registry studies are mainly required for drugs where substantial use during pregnancy is foreseen, for example, in malaria or for HIV treatment, monitoring pregnancy outcomes in women exposed to drugs during gestation (Postmarket_Requirements_and_Commitments_[fda.gov]). In addition to data collection in registries, when pregnancy is expected to impact systemic drug levels, clinical PK data are generated post-authorization to inform dosing recommendations (e.g., rilpivirine, darunavir, cobicistat).</p><p>For years, the clinical need for drug treatment during pregnancy has been left largely unresolved by regulators and sponsors, leaving the risk–benefit assessment to prescribers and patients. However, health care and community attention to this unmet medical need has resulted in increased regulatory action. In 2018, the FDA published a draft guidance on Scientific and Ethical Considerations for the Inclusion of pregnant women in Clinical Trials. In 2022, current thinking and regulatory efforts were communicated by Sewell et al.<span><sup>2</sup></span> and the FDA diversity plan framework (Diversity_Plans_to_Improve_Enrollment_of_Participants_from_Underrepresented_Racial_and_Ethnic_Populations_in_Clinical_Trials_Guidance_for_Industry_(fda.gov)). Furthermore, regulators from the FDA, EMA, and MHRA acknowledged the urgent need to shift from systematic exclusion to the inclusion of pregnant and breastfeeding women in clinical trials at the International Coalition of Medicines Regulatory Authorities Pregnancy and Lactation Workshop (ICMRA_Pregnancy_and_Lactation_Workshop_International_Coalition_of_Medicines_Regulatory_Authorities_(ICMRA)). It was proposed that applicants should develop and submit a “Maternal Investigation Plan,” outlining the strategy to study these populations. This change of approach requires international collaboration and harmonization, and foundations were laid for the development of the ICH21 Guideline (ICH_E21_Final_Concept_Paper_2023) which will outline the investigational development plan, alongside other factors considered in the extrapolation framework of the draft ICH E11 guideline for pediatrics (draft-ich-guideline-e11a-pediatric-extrapolation-step-2b_en.pdf_(europa.eu)). The FDA has also held workshops to encourage an increase in the number of studies conducted in pregnant woman (Pharmacokinetic_Evaluation_in_Pregnancy_(fda.gov); Fetal_Pharmacology_and_Therapeutics_(fda.gov)).</p><p>Pregnancy-related physiological changes can affect drug PK resulting in possible loss of efficacy, or potential toxicity in both the mother and fetus. Phase I (e.g., CYP3A4, CYP2D6, and CYP219) and phase II (UGT1A1 and UGT1A4) metabolizing enzyme activities are altered during pregnancy. This is illustrated by the observed ~60% increase in clearance and ~60% decrease in exposure of the CYP2D6 substrates metoprolol and fluoxetine, respectively.<span><sup>3, 4</sup></span> Corresponding decreases in exposure have been reported for the CYP3A substrates rilpivirine and cobicistat-boosted darunavir as well as for the UGT1A1 substrate dolutegravir. Increased glomerular filtration rate during pregnancy, can lead to reduced exposure of drugs undergoing renal excretion (e.g., ceftazidime).</p><p>In some cases, the decrease in exposure has been reflected in the labeling, driving either dose recommendations, monitoring, or contraindications.</p><p>In the original rilpivirine label (2011), the use of the drug during pregnancy was restricted to cases where the potential benefit justifies the potential risk. In 2018, the labeling was updated based on new data (i.e., 30%–40% lower exposure in second and third trimesters than postpartum), resulting in a recommendation to closely monitor viral load due to lower exposures during pregnancy (EDURANT®_(rilpivirine)_US_label).</p><p>Similarly, the exposure of dolutegravir was reported to be up to 37% lower during pregnancy.<span><sup>5</sup></span> In the recently updated label (2024), clinical fetal outcome data from observational studies supported the removal of pregnancy testing before initiation of treatment as well as the warning of embryo–fetal toxicity (TIVICAY_(dolutegravir)_US_label).</p><p>As an example of major labeling impact, the exposure of darunavir boosted with cobicistat was substantially lower (>50% decrease in AUC, and ~90% decrease in <i>C</i><sub>min</sub>) during the second and third trimesters. Hence, the label states that use during pregnancy is not recommended. An exposure registry monitors pregnancy outcomes in individuals taking the drugs (PREZCOBIX®_(darunavir_and_cobicistat)_US_label). Publicly available data are not sufficient to allow the evaluation of whether a dose increase may be feasible and safe for the mother and fetus.</p><p>The EMA recommends therapeutic drug monitoring for ritonavir-boosted atazanavir (300/100 mg) as insufficient exposure to atazanavir may occur in pregnant patients. Moreover, both EMA and FDA labels state that if either tenofovir or an H2-receptor antagonist is co-administered in pregnant women, an atazanavir dose increase (400 mg) may be required due to enhanced exposure reduction (REYATAZ_[atazanavir]_US_label; REYATAZ_EMA_SmPC).</p><p>Before these labeling changes were implemented, it is likely that uninformed, off-label use occurred.</p><p>The pediatric extrapolation framework outlined in ICH E11 (ICH_Guideline_E11A_on_Pediatric_Extrapolation) can be adapted to pregnancy, supporting decisions on the need for studies, informing study design and labeling update, and generating new data to fill the knowledge gaps. Particular consideration should be made for teratogenicity and fetal toxicity, and a cautious, informed, approach is recommended. Evaluation and interpretation of fetal and preclinical data informing risk should be considered but it is outside the scope of this paper. Registries provide important follow-up information on fetal, that is, pediatric safety detecting signals and allow broader scientific analysis. If the use of a medicine in pregnant women, during certain trimesters, is too rare for conventional studies to be possible, adapted strategies to obtain PK and clinical information should be planned for. Labeling language could be considered to enable these approaches.</p><p>As in ICH E11, the drug development plan should consider the clinical need for treatment and foreseen risks in each trimester as well as significant benefits compared with available therapies. The efficacy extrapolation approach could be based on similarity in disease and in response to treatment, drug pharmacology, expected similarity in target exposure, and how to reach this exposure in pregnant individuals. Scientific literature, preclinical and clinical data, clinical experience with other drugs, including RWE, and foreseen PK differences, should all be considered (Figure 1).</p><p>The extrapolation framework could support decisions on mother and fetal safety management including study design, sample size (which might be smaller than conventional phase III), generation of post-authorization safety data in a larger population, need for long-term follow-up and registries, etc. Key considerations also include risks to mother and fetus if not treating the disease, the need for gradual recruitment into earlier trimesters, available safety information in nonpregnant women, whether target/off-target effects could be different, or particularly impact pregnant women or the fetus. Safety data available for drugs with similar mechanisms of action, or off-target effects are crucial, as well as preclinical fetal toxicity/teratogenicity information and the need to follow-up specific short- and long-term effects, etc. (Table 1). Expected fetal exposure to drugs should if possible be considered.</p><p>Availability of data assessing similarity of exposure–response (ER) relationships for efficacy and safety compared with nonpregnant should be reflected in the study design, and physiological factors impacting drug exposure during pregnancy considered in dose selection, and significant uncertainties resolved by confirmatory “PK lead-in” investigations.</p><p>Sparse sampling approaches should be optimized using population PK (pop-PK) to support study designs, for example, informing sample size estimations. The use of physiologically-based pharmacokinetic (PBPK) modeling to simulate drug exposure is recommended for initial dose selection, as it allows multifactorial mechanistic knowledge to inform exposure predictions. For marketed drugs, the analysis of pharmacovigilance and real-world data on use during pregnancy should be considered. It is also important to understand whether loss of efficacy is expected due to altered drug systemic exposure, and if temporary dose adjustment is needed.</p><p>The clinical pharmacology strategy for investigating the impact of pregnancy on drug exposure requires integration of available data, including clinical PK data collected in nonpregnant populations, preclinical information, a good understanding of the effect of pregnancy-related physiological changes on drug exposure, potential DDIs, and exposure–response relationship. The totality of evidence can inform the development of a MIDD-based extrapolation framework to identify and fill knowledge gaps underlining a development strategy to optimize the use of available data.</p><p>The use of PBPK models, incorporating relevant physiological changes, to predict drug exposure and PD response (target and off-target pharmacological effects) in pregnant women and the fetus is being considered by regulators as a tool to possibly inform dosing during pregnancy and support benefit–risk decisions [ICH_E21_Final_Concept_Paper_2023].<span><sup>6, 7</sup></span></p><p>When there is a good understanding of the effect of pregnancy on the physiological factors impacting the disposition of the specific drug, PBPK modeling can be confidently applied to predict exposure changes and drive dosing decisions. Several authors showed that PBPK modeling can reliably predict those changes in exposure in pregnancy compared with nonpregnant populations for a range of drugs,<span><sup>8, 9</sup></span> [Pregnancy_Physiologically_Based_Pharmacokinetic_(PBPK)_Modeling_with_Population_Variability_for_Drug_Safety_and_Efficacy_Assessment_(fda.gov)]. A high-fidelity drug model<span><sup>10</sup></span> (where the elimination routes are clearly elucidated) developed initially in the nonpregnant population can then be applied to predict an initial dose in pregnant subjects. Once PK data in the pregnant patients have been accrued, pop-PK modeling could be used to support the PBPK modeling, quantify ER relationships, and analyze covariates affecting exposure.</p><p>A change of mindset appears to have been adopted by the scientific and regulatory community. The need for pregnant women to have well-informed medical treatments has been recognized, going from a protection from research to a protection by research strategy. The risk of fetal toxicity should be reflected in relevant parts of the development and multifactorial risk–benefit discussions. The regulatory framework could be based on approaches like global pediatric regulations. Indeed, we propose to adapt the pediatric development extrapolation framework, (ICH_Guideline_E11A_on_Pediatric_Extrapolation), providing a systematic approach to drug development, identifying important knowledge gaps to be addressed in the clinical study design and follow-up strategies. A clinical pharmacology strategy based on MIDD approaches, such as PBPK and pop-PK is central to this approach.</p><p>No funding was received for this work.</p><p>P.C. is an employee of Certara Italy (Certara Drug Development Solutions). E.B. is an employee of Certara NL (Certara Drug Development Solutions). K.R.Y. is an employee of Certara UK Limited (Simcyp Division).</p><p>As Deputy-Editor-Chief for <i>CPT: Pharmacometrics & Systems Pharmacology</i>, Karen Rowland Yeo was not involved in the review or decision-making processes for this work.</p>\",\"PeriodicalId\":10774,\"journal\":{\"name\":\"CPT: Pharmacometrics & Systems Pharmacology\",\"volume\":\"13 11\",\"pages\":\"1830-1834\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-10-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/psp4.13242\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"CPT: Pharmacometrics & Systems Pharmacology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/psp4.13242\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"PHARMACOLOGY & PHARMACY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"CPT: Pharmacometrics & Systems Pharmacology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/psp4.13242","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
Medicines in pregnancy: A clinical pharmacology extrapolation framework to address knowledge gaps
Drug treatment may be required during pregnancy, both for pregnant women and their unborn children. About 6 million pregnancies in the United States (US) occur each year, with most women taking at least one prescription medication during pregnancy and more than half of the mothers taking medicines after delivery (Pregnant?_Breastfeeding?_FDA_Aims_to_Improve_Drug_Information_[fda.gov]). However, in our attempts to protect the unborn children or breastfeeding infants, information to support such treatment is rarely generated and drugs are often used off-label.
Systematic exclusion of pregnant women from clinical trials at all stages does not allow the collection of data to support the safe use of medicines during pregnancy. Dosing strategies to treat health conditions developed either before or during pregnancy often rely on data from healthy and/or nonpregnant subjects, instead of being driven by complex pregnancy-related physiological changes on drug exposure. Despite the recognized medical need, a recent review captured labels with clinically meaningful interventions in pregnancy for only 139 medications in the US and 20 in the European Union (EU); in both cases, 30%–40% had established doses for pregnant populations.1 Information on dosing during pregnancy is often unavailable in original regulatory submissions limiting label recommendation. Pre-authorization data in pregnant population is generally not requested and post-authorization registry studies are mainly required for drugs where substantial use during pregnancy is foreseen, for example, in malaria or for HIV treatment, monitoring pregnancy outcomes in women exposed to drugs during gestation (Postmarket_Requirements_and_Commitments_[fda.gov]). In addition to data collection in registries, when pregnancy is expected to impact systemic drug levels, clinical PK data are generated post-authorization to inform dosing recommendations (e.g., rilpivirine, darunavir, cobicistat).
For years, the clinical need for drug treatment during pregnancy has been left largely unresolved by regulators and sponsors, leaving the risk–benefit assessment to prescribers and patients. However, health care and community attention to this unmet medical need has resulted in increased regulatory action. In 2018, the FDA published a draft guidance on Scientific and Ethical Considerations for the Inclusion of pregnant women in Clinical Trials. In 2022, current thinking and regulatory efforts were communicated by Sewell et al.2 and the FDA diversity plan framework (Diversity_Plans_to_Improve_Enrollment_of_Participants_from_Underrepresented_Racial_and_Ethnic_Populations_in_Clinical_Trials_Guidance_for_Industry_(fda.gov)). Furthermore, regulators from the FDA, EMA, and MHRA acknowledged the urgent need to shift from systematic exclusion to the inclusion of pregnant and breastfeeding women in clinical trials at the International Coalition of Medicines Regulatory Authorities Pregnancy and Lactation Workshop (ICMRA_Pregnancy_and_Lactation_Workshop_International_Coalition_of_Medicines_Regulatory_Authorities_(ICMRA)). It was proposed that applicants should develop and submit a “Maternal Investigation Plan,” outlining the strategy to study these populations. This change of approach requires international collaboration and harmonization, and foundations were laid for the development of the ICH21 Guideline (ICH_E21_Final_Concept_Paper_2023) which will outline the investigational development plan, alongside other factors considered in the extrapolation framework of the draft ICH E11 guideline for pediatrics (draft-ich-guideline-e11a-pediatric-extrapolation-step-2b_en.pdf_(europa.eu)). The FDA has also held workshops to encourage an increase in the number of studies conducted in pregnant woman (Pharmacokinetic_Evaluation_in_Pregnancy_(fda.gov); Fetal_Pharmacology_and_Therapeutics_(fda.gov)).
Pregnancy-related physiological changes can affect drug PK resulting in possible loss of efficacy, or potential toxicity in both the mother and fetus. Phase I (e.g., CYP3A4, CYP2D6, and CYP219) and phase II (UGT1A1 and UGT1A4) metabolizing enzyme activities are altered during pregnancy. This is illustrated by the observed ~60% increase in clearance and ~60% decrease in exposure of the CYP2D6 substrates metoprolol and fluoxetine, respectively.3, 4 Corresponding decreases in exposure have been reported for the CYP3A substrates rilpivirine and cobicistat-boosted darunavir as well as for the UGT1A1 substrate dolutegravir. Increased glomerular filtration rate during pregnancy, can lead to reduced exposure of drugs undergoing renal excretion (e.g., ceftazidime).
In some cases, the decrease in exposure has been reflected in the labeling, driving either dose recommendations, monitoring, or contraindications.
In the original rilpivirine label (2011), the use of the drug during pregnancy was restricted to cases where the potential benefit justifies the potential risk. In 2018, the labeling was updated based on new data (i.e., 30%–40% lower exposure in second and third trimesters than postpartum), resulting in a recommendation to closely monitor viral load due to lower exposures during pregnancy (EDURANT®_(rilpivirine)_US_label).
Similarly, the exposure of dolutegravir was reported to be up to 37% lower during pregnancy.5 In the recently updated label (2024), clinical fetal outcome data from observational studies supported the removal of pregnancy testing before initiation of treatment as well as the warning of embryo–fetal toxicity (TIVICAY_(dolutegravir)_US_label).
As an example of major labeling impact, the exposure of darunavir boosted with cobicistat was substantially lower (>50% decrease in AUC, and ~90% decrease in Cmin) during the second and third trimesters. Hence, the label states that use during pregnancy is not recommended. An exposure registry monitors pregnancy outcomes in individuals taking the drugs (PREZCOBIX®_(darunavir_and_cobicistat)_US_label). Publicly available data are not sufficient to allow the evaluation of whether a dose increase may be feasible and safe for the mother and fetus.
The EMA recommends therapeutic drug monitoring for ritonavir-boosted atazanavir (300/100 mg) as insufficient exposure to atazanavir may occur in pregnant patients. Moreover, both EMA and FDA labels state that if either tenofovir or an H2-receptor antagonist is co-administered in pregnant women, an atazanavir dose increase (400 mg) may be required due to enhanced exposure reduction (REYATAZ_[atazanavir]_US_label; REYATAZ_EMA_SmPC).
Before these labeling changes were implemented, it is likely that uninformed, off-label use occurred.
The pediatric extrapolation framework outlined in ICH E11 (ICH_Guideline_E11A_on_Pediatric_Extrapolation) can be adapted to pregnancy, supporting decisions on the need for studies, informing study design and labeling update, and generating new data to fill the knowledge gaps. Particular consideration should be made for teratogenicity and fetal toxicity, and a cautious, informed, approach is recommended. Evaluation and interpretation of fetal and preclinical data informing risk should be considered but it is outside the scope of this paper. Registries provide important follow-up information on fetal, that is, pediatric safety detecting signals and allow broader scientific analysis. If the use of a medicine in pregnant women, during certain trimesters, is too rare for conventional studies to be possible, adapted strategies to obtain PK and clinical information should be planned for. Labeling language could be considered to enable these approaches.
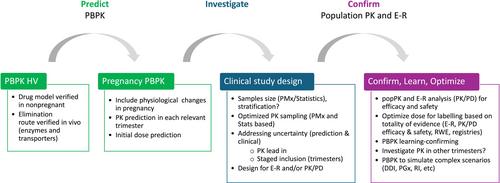
As in ICH E11, the drug development plan should consider the clinical need for treatment and foreseen risks in each trimester as well as significant benefits compared with available therapies. The efficacy extrapolation approach could be based on similarity in disease and in response to treatment, drug pharmacology, expected similarity in target exposure, and how to reach this exposure in pregnant individuals. Scientific literature, preclinical and clinical data, clinical experience with other drugs, including RWE, and foreseen PK differences, should all be considered (Figure 1).
The extrapolation framework could support decisions on mother and fetal safety management including study design, sample size (which might be smaller than conventional phase III), generation of post-authorization safety data in a larger population, need for long-term follow-up and registries, etc. Key considerations also include risks to mother and fetus if not treating the disease, the need for gradual recruitment into earlier trimesters, available safety information in nonpregnant women, whether target/off-target effects could be different, or particularly impact pregnant women or the fetus. Safety data available for drugs with similar mechanisms of action, or off-target effects are crucial, as well as preclinical fetal toxicity/teratogenicity information and the need to follow-up specific short- and long-term effects, etc. (Table 1). Expected fetal exposure to drugs should if possible be considered.
Availability of data assessing similarity of exposure–response (ER) relationships for efficacy and safety compared with nonpregnant should be reflected in the study design, and physiological factors impacting drug exposure during pregnancy considered in dose selection, and significant uncertainties resolved by confirmatory “PK lead-in” investigations.
Sparse sampling approaches should be optimized using population PK (pop-PK) to support study designs, for example, informing sample size estimations. The use of physiologically-based pharmacokinetic (PBPK) modeling to simulate drug exposure is recommended for initial dose selection, as it allows multifactorial mechanistic knowledge to inform exposure predictions. For marketed drugs, the analysis of pharmacovigilance and real-world data on use during pregnancy should be considered. It is also important to understand whether loss of efficacy is expected due to altered drug systemic exposure, and if temporary dose adjustment is needed.
The clinical pharmacology strategy for investigating the impact of pregnancy on drug exposure requires integration of available data, including clinical PK data collected in nonpregnant populations, preclinical information, a good understanding of the effect of pregnancy-related physiological changes on drug exposure, potential DDIs, and exposure–response relationship. The totality of evidence can inform the development of a MIDD-based extrapolation framework to identify and fill knowledge gaps underlining a development strategy to optimize the use of available data.
The use of PBPK models, incorporating relevant physiological changes, to predict drug exposure and PD response (target and off-target pharmacological effects) in pregnant women and the fetus is being considered by regulators as a tool to possibly inform dosing during pregnancy and support benefit–risk decisions [ICH_E21_Final_Concept_Paper_2023].6, 7
When there is a good understanding of the effect of pregnancy on the physiological factors impacting the disposition of the specific drug, PBPK modeling can be confidently applied to predict exposure changes and drive dosing decisions. Several authors showed that PBPK modeling can reliably predict those changes in exposure in pregnancy compared with nonpregnant populations for a range of drugs,8, 9 [Pregnancy_Physiologically_Based_Pharmacokinetic_(PBPK)_Modeling_with_Population_Variability_for_Drug_Safety_and_Efficacy_Assessment_(fda.gov)]. A high-fidelity drug model10 (where the elimination routes are clearly elucidated) developed initially in the nonpregnant population can then be applied to predict an initial dose in pregnant subjects. Once PK data in the pregnant patients have been accrued, pop-PK modeling could be used to support the PBPK modeling, quantify ER relationships, and analyze covariates affecting exposure.
A change of mindset appears to have been adopted by the scientific and regulatory community. The need for pregnant women to have well-informed medical treatments has been recognized, going from a protection from research to a protection by research strategy. The risk of fetal toxicity should be reflected in relevant parts of the development and multifactorial risk–benefit discussions. The regulatory framework could be based on approaches like global pediatric regulations. Indeed, we propose to adapt the pediatric development extrapolation framework, (ICH_Guideline_E11A_on_Pediatric_Extrapolation), providing a systematic approach to drug development, identifying important knowledge gaps to be addressed in the clinical study design and follow-up strategies. A clinical pharmacology strategy based on MIDD approaches, such as PBPK and pop-PK is central to this approach.
No funding was received for this work.
P.C. is an employee of Certara Italy (Certara Drug Development Solutions). E.B. is an employee of Certara NL (Certara Drug Development Solutions). K.R.Y. is an employee of Certara UK Limited (Simcyp Division).
As Deputy-Editor-Chief for CPT: Pharmacometrics & Systems Pharmacology, Karen Rowland Yeo was not involved in the review or decision-making processes for this work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


