利用广义核电子轨道多态密度泛函理论研究多质子转移过程的非绝热氢隧道动力学
IF 5.7
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
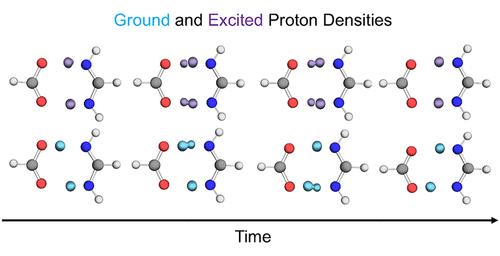
质子转移和氢隧道在许多重要的化学和生物过程中发挥着关键作用。为了捕捉涉及一个或多个质子转移和隧穿的系统中的氢隧穿效应,我们开发了广义核电子轨道多态密度泛函理论(NEO-MSDFT)方法。广义 NEO-MSDFT 方法在与电子相同的水平上对转移质子进行量子力学处理,并通过在非正交构型相互作用方案中混合局部 NEO-DFT 态,获得与氢隧道相关的脱局域振子态。在这里,我们介绍了广义 NEO-MSDFT 振子态能量和这些振子态之间的非绝热耦合向量的分析梯度的推导和实现。我们使用这种方法对甲酸二聚体以及甲脒和甲酸的异二聚体中的双质子转移反应进行绝热和非绝热动力学模拟。结果表明,广义 NEO-MSDFT 方法可以捕捉到这些过程中两个质子的强耦合同步或异步隧道。研究发现,加入振动非绝热效应会对双质子转移动力学产生重大影响。这项工作为质子中继和氢键网络等多质子传递系统的各种非绝热动力学模拟奠定了基础。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Nonadiabatic Hydrogen Tunneling Dynamics for Multiple Proton Transfer Processes with Generalized Nuclear-Electronic Orbital Multistate Density Functional Theory
Proton transfer and hydrogen tunneling play key roles in many processes of chemical and biological importance. The generalized nuclear-electronic orbital multistate density functional theory (NEO-MSDFT) method was developed in order to capture hydrogen tunneling effects in systems involving the transfer and tunneling of one or more protons. The generalized NEO-MSDFT method treats the transferring protons quantum mechanically on the same level as the electrons and obtains the delocalized vibronic states associated with hydrogen tunneling by mixing localized NEO-DFT states in a nonorthogonal configuration interaction scheme. Herein, we present the derivation and implementation of analytical gradients for the generalized NEO-MSDFT vibronic state energies and the nonadiabatic coupling vectors between these vibronic states. We use this methodology to perform adiabatic and nonadiabatic dynamics simulations of the double proton transfer reactions in the formic acid dimer and the heterodimer of formamidine and formic acid. The generalized NEO-MSDFT method is shown to capture the strongly coupled synchronous or asynchronous tunneling of the two protons in these processes. Inclusion of vibronically nonadiabatic effects is found to significantly impact the double proton transfer dynamics. This work lays the foundation for a variety of nonadiabatic dynamics simulations of multiple proton transfer systems, such as proton relays and hydrogen-bonding networks.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


