Richard Schier, Daniel Guo, Holger-Dietrich Saßnick, Caterina Cocchi
{"title":"高通量 Ab Initio 计算得出的 K-Sb 和 Na-Sb 二元晶体的稳定性和电子特性","authors":"Richard Schier, Daniel Guo, Holger-Dietrich Saßnick, Caterina Cocchi","doi":"10.1002/adts.202400680","DOIUrl":null,"url":null,"abstract":"<p>The study of the fundamental properties of alkali antimonide photocathodes for particle accelerators is currently hindered by the limited purity of the samples. First-principles studies can effectively complement experiments to gain insight into the stability and the electronic structure of these compounds. In this high-throughput analysis based on density-functional theory (DFT), two families of binary crystals with K-Sb and Na-Sb compositions expected to form during evaporation of multi-alkali antimonide photocathodes are investigated. Starting from an initial pool of structures mined from existing computational databases, automatized routines included in the in-house developed library <span>aim<sup>2</sup>dat</span> are employed to determine the stability and the electronic properties of the aforementioned systems. By analyzing the formation energy, the structures are ranked in a convex hull retaining the information of their crystalline arrangement. Next, the band structure and the projected density of states of selected stable compounds are analyzed. Adopting the r<sup>2</sup>SCAN functional for the DFT calculations, reliable estimates of the character and size of the bandgaps are obtained and discussed in relation to the relative alkali content in the crystals. These results provide useful indications to predict and characterize binary phases forming during the growth of multi-alkali antimonide photocathodes.</p>","PeriodicalId":7219,"journal":{"name":"Advanced Theory and Simulations","volume":"7 12","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2024-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/adts.202400680","citationCount":"0","resultStr":"{\"title\":\"Stability and Electronic Properties of K-Sb and Na-Sb Binary Crystals from High-Throughput Ab Initio Calculations\",\"authors\":\"Richard Schier, Daniel Guo, Holger-Dietrich Saßnick, Caterina Cocchi\",\"doi\":\"10.1002/adts.202400680\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The study of the fundamental properties of alkali antimonide photocathodes for particle accelerators is currently hindered by the limited purity of the samples. First-principles studies can effectively complement experiments to gain insight into the stability and the electronic structure of these compounds. In this high-throughput analysis based on density-functional theory (DFT), two families of binary crystals with K-Sb and Na-Sb compositions expected to form during evaporation of multi-alkali antimonide photocathodes are investigated. Starting from an initial pool of structures mined from existing computational databases, automatized routines included in the in-house developed library <span>aim<sup>2</sup>dat</span> are employed to determine the stability and the electronic properties of the aforementioned systems. By analyzing the formation energy, the structures are ranked in a convex hull retaining the information of their crystalline arrangement. Next, the band structure and the projected density of states of selected stable compounds are analyzed. Adopting the r<sup>2</sup>SCAN functional for the DFT calculations, reliable estimates of the character and size of the bandgaps are obtained and discussed in relation to the relative alkali content in the crystals. These results provide useful indications to predict and characterize binary phases forming during the growth of multi-alkali antimonide photocathodes.</p>\",\"PeriodicalId\":7219,\"journal\":{\"name\":\"Advanced Theory and Simulations\",\"volume\":\"7 12\",\"pages\":\"\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-09-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/adts.202400680\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Advanced Theory and Simulations\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/adts.202400680\",\"RegionNum\":4,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advanced Theory and Simulations","FirstCategoryId":"5","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/adts.202400680","RegionNum":4,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
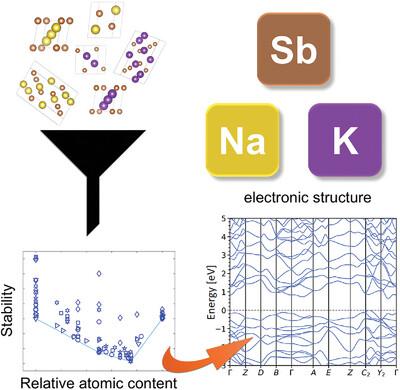
Stability and Electronic Properties of K-Sb and Na-Sb Binary Crystals from High-Throughput Ab Initio Calculations
The study of the fundamental properties of alkali antimonide photocathodes for particle accelerators is currently hindered by the limited purity of the samples. First-principles studies can effectively complement experiments to gain insight into the stability and the electronic structure of these compounds. In this high-throughput analysis based on density-functional theory (DFT), two families of binary crystals with K-Sb and Na-Sb compositions expected to form during evaporation of multi-alkali antimonide photocathodes are investigated. Starting from an initial pool of structures mined from existing computational databases, automatized routines included in the in-house developed library aim2dat are employed to determine the stability and the electronic properties of the aforementioned systems. By analyzing the formation energy, the structures are ranked in a convex hull retaining the information of their crystalline arrangement. Next, the band structure and the projected density of states of selected stable compounds are analyzed. Adopting the r2SCAN functional for the DFT calculations, reliable estimates of the character and size of the bandgaps are obtained and discussed in relation to the relative alkali content in the crystals. These results provide useful indications to predict and characterize binary phases forming during the growth of multi-alkali antimonide photocathodes.
期刊介绍:
Advanced Theory and Simulations is an interdisciplinary, international, English-language journal that publishes high-quality scientific results focusing on the development and application of theoretical methods, modeling and simulation approaches in all natural science and medicine areas, including:
materials, chemistry, condensed matter physics
engineering, energy
life science, biology, medicine
atmospheric/environmental science, climate science
planetary science, astronomy, cosmology
method development, numerical methods, statistics


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


