Nichole A Lewis, Rachel Herndon Klein, Cailin Kelly, Jennifer Yee, Paul S Knoepfler
{"title":"Histone H3.3 K27M chromatin functions implicate a network of neurodevelopmental factors including ASCL1 and NEUROD1 in DIPG.","authors":"Nichole A Lewis, Rachel Herndon Klein, Cailin Kelly, Jennifer Yee, Paul S Knoepfler","doi":"10.1186/s13072-022-00447-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The histone variant H3.3 K27M mutation is a defining characteristic of diffuse intrinsic pontine glioma (DIPG)/diffuse midline glioma (DMG). This histone mutation is responsible for major alterations to histone H3 post-translational modification (PTMs) and subsequent aberrant gene expression. However, much less is known about the effect this mutation has on chromatin structure and function, including open versus closed chromatin regions as well as their transcriptomic consequences.</p><p><strong>Results: </strong>Recently, we developed isogenic CRISPR-edited DIPG cell lines that are wild-type for histone H3.3 that can be compared to their matched K27M lines. Here we show via ATAC-seq analysis that H3.3K27M glioma cells have unique accessible chromatin at regions corresponding to neurogenesis, NOTCH, and neuronal development pathways and associated genes that are overexpressed in H3.3K27M compared to our isogenic wild-type cell line. As to mechanisms, accessible enhancers and super-enhancers corresponding to increased gene expression in H3.3K27M cells were also mapped to genes involved in neurogenesis and NOTCH signaling, suggesting that these pathways are key to DIPG tumor maintenance. Motif analysis implicates specific transcription factors as central to the neuro-oncogenic K27M signaling pathway, in particular, ASCL1 and NEUROD1.</p><p><strong>Conclusions: </strong>Altogether our findings indicate that H3.3K27M causes chromatin to take on a more accessible configuration at key regulatory regions for NOTCH and neurogenesis genes resulting in increased oncogenic gene expression, which is at least partially reversible upon editing K27M back to wild-type.</p>","PeriodicalId":49253,"journal":{"name":"Epigenetics & Chromatin","volume":"15 1","pages":"18"},"PeriodicalIF":4.2000,"publicationDate":"2022-05-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9121554/pdf/","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Epigenetics & Chromatin","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13072-022-00447-6","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 6
Abstract
Background: The histone variant H3.3 K27M mutation is a defining characteristic of diffuse intrinsic pontine glioma (DIPG)/diffuse midline glioma (DMG). This histone mutation is responsible for major alterations to histone H3 post-translational modification (PTMs) and subsequent aberrant gene expression. However, much less is known about the effect this mutation has on chromatin structure and function, including open versus closed chromatin regions as well as their transcriptomic consequences.
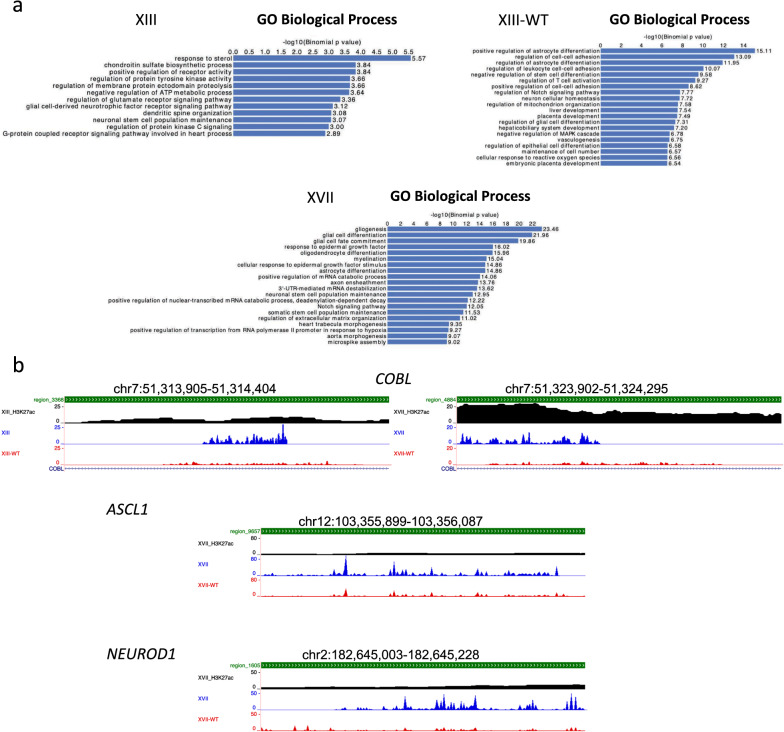
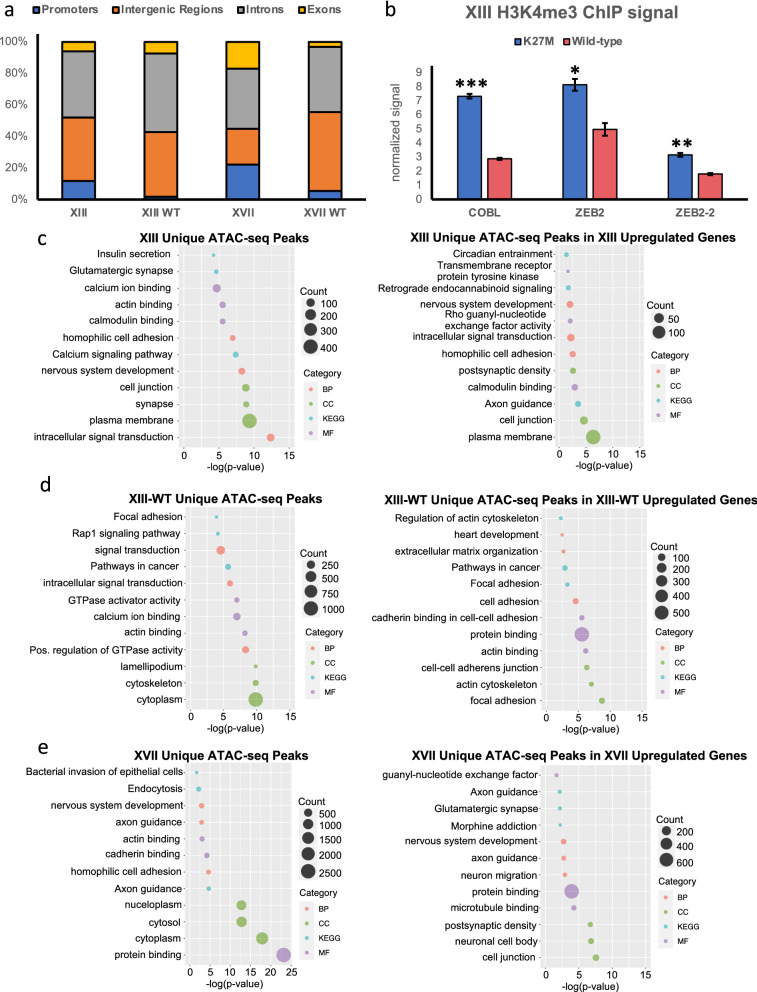
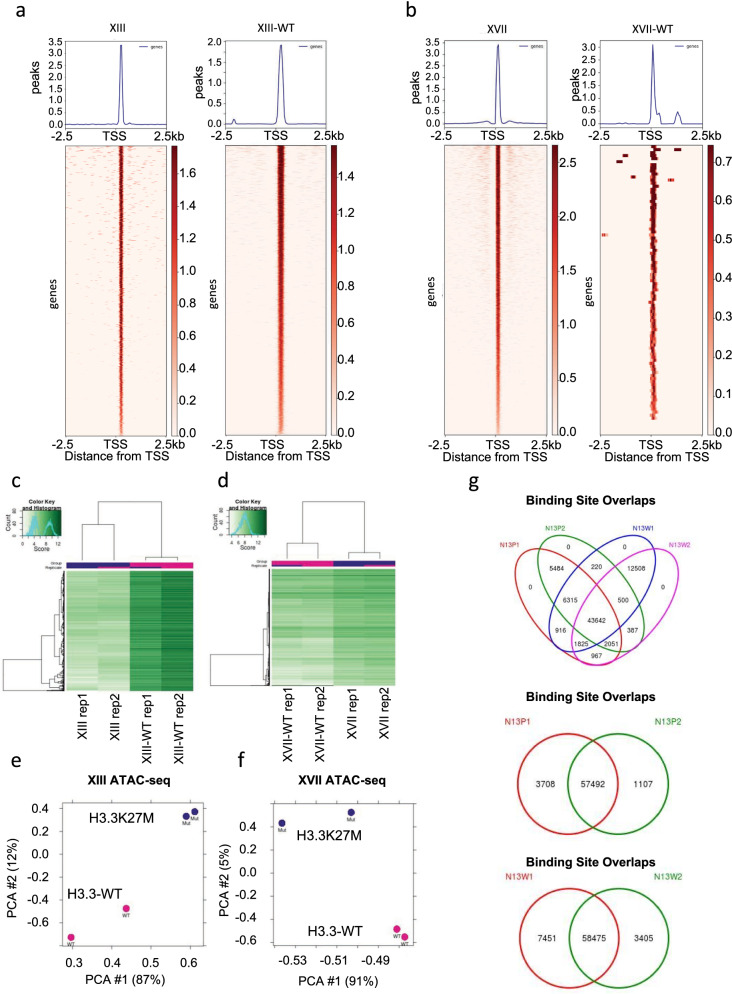
Results: Recently, we developed isogenic CRISPR-edited DIPG cell lines that are wild-type for histone H3.3 that can be compared to their matched K27M lines. Here we show via ATAC-seq analysis that H3.3K27M glioma cells have unique accessible chromatin at regions corresponding to neurogenesis, NOTCH, and neuronal development pathways and associated genes that are overexpressed in H3.3K27M compared to our isogenic wild-type cell line. As to mechanisms, accessible enhancers and super-enhancers corresponding to increased gene expression in H3.3K27M cells were also mapped to genes involved in neurogenesis and NOTCH signaling, suggesting that these pathways are key to DIPG tumor maintenance. Motif analysis implicates specific transcription factors as central to the neuro-oncogenic K27M signaling pathway, in particular, ASCL1 and NEUROD1.
Conclusions: Altogether our findings indicate that H3.3K27M causes chromatin to take on a more accessible configuration at key regulatory regions for NOTCH and neurogenesis genes resulting in increased oncogenic gene expression, which is at least partially reversible upon editing K27M back to wild-type.
期刊介绍:
Epigenetics & Chromatin is a peer-reviewed, open access, online journal that publishes research, and reviews, providing novel insights into epigenetic inheritance and chromatin-based interactions. The journal aims to understand how gene and chromosomal elements are regulated and their activities maintained during processes such as cell division, differentiation and environmental alteration.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


