Correction to “Ab Initio Investigation of CH4 Dehydrogenation on a (CeO2)10 Cluster”
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
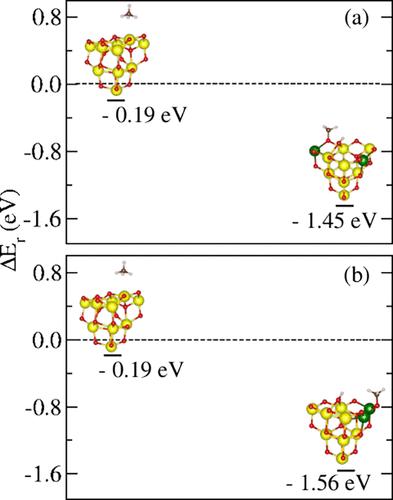
Unfortunately, we identified two errors in our paper related to the misapplication of the unity bond index–quadratic exponential potential (UBI–QEP) analysis, which became evident through discussions with Dr. Verónica Ganduglia-Pirovano and Dr. Breno L. Galvão (Private communication, March 15, 2024). The first error involves the results used to design Figure 5, where we obtained the activation energy values using the UBI-QEP approximation. In this analysis, we considered the most common adsorption sites reported in the literature for the CeO2(111) surface rather than the most stable ones on the ceria cluster. As a result, the dependence between the CH4 adsorption site and the corresponding CH3 and/or H sites was not maintained. A more appropriate approach for our purposes would be to conduct a systematic investigation of the coadsorption sites on (CeO2)10, using a specific adsorption site as a reference and mapping the most stable CH3 or H sites. Therefore, we implemented the aforementioned approach by mapping the CH3 sites and fixing the most stable H site as a reference; hence, the text and the related figure (Figure 5) on page 11944 should be updated based on the obtained new results. The second error concerns the formulation of Figure 6, where the transverse lines connecting the structures may give the reader the mistaken impression of a proposed reaction path. In reality, we present the reaction energy values for representative structures of each group selected by the k-means clustering method. Regarding the mistake in the production of Figures 5 and 6, the following changes must be applied: On page 11946, the sentence “The CH4 molecule can adsorb on the nanocluster considering four adsorption configurations, [...]” must be replaced with “The CH4 molecule can adsorb on the nanocluster considering three adsorption configurations, [...]” and the sentence “[...] C–H bond break on the (CeO2)10 nanocluster can be related to the H adsorption site [...]” must be replaced with “[...] C–H bond break on the (CeO2)10 nanocluster can be related to the coadsorption sites of CH3 (H)[...]”. The original Figure 5 should be replaced by Figure 5 of this Correction. On page 11944, the sentence “We found four different adsorption modes for CH4 on (CeO2)10, namely, umbrella, antiumbrella, scissoring, and modified antiumbrella, as indicated in Figure 5.66 In the umbrella and antiumbrella modes, the calculated barrier (Ea) for the CH4 dissociation (CH4 → CH3 + H*) indicates a higher cluster reactivity compared to the CeO2(111) surface. In this case, we obtained Ea equal to 0.54 eV (EaZPE = 0.48 eV) and 0.59 eV (EaZPE = 0.52 eV) for umbrella and antiumbrella modes, respectively, smaller than the value reported for the CeO2(111) surface (1.44 eV).65” must be replaced with “We found three different adsorption modes for CH4 on (CeO2)10 clusters, namely umbrella, antiumbrella, and scissoring.66 The calculated barrier (Ea) for the CH4 dissociation (CH4 → CH3 + H*) indicates a higher cluster reactivity compared to the CeO2(111) surface. In this case, we obtained Ea equal to 0.07 eV (EaZPE = 0.03) and 0.13 eV (EaZPE = 0.09 eV) for the configuration proposed in Figure 1a,b, respectively, smaller than the value reported for the CeO2(111) surface (1.44 eV).65” In page 11944, the sentence “Then, the adsorption energy for the umbrella and antiumbrella modes in this state is −3.59 and −3.95 eV, respectively, in agreement with the value of −3.68 eV reported for CeO2(111) surface.25” must be removed. The original Figure 6 should be replaced by Figure 6 of this Correction. On page 11945, the following paragraph must be removed: “The energy barriers for the modified antiumbrella and scissoring modes (0.81 and 0.64 eV, respectively) indicate a higher barrier compared to the umbrella and antiumbrella modes, which can be associated with the H adsorption site. Once again, the ZPE corrections do not produce significant changes in the Ea values (0.74 and 0.56 eV, respectively). The difference between the modified and antiumbrella modes is the H adsorption site, since the former adsorbs in the 3-fold topO site, allowing the molecule adsorption at a 2-fold topO site. The opposite occurs for the latter one, in which the molecule adsorbs on the adsorption O site with a higher coordination. However, the activation energy obtained for the modified antiumbrella and scissoring mode can be compared with the values reported by Righi et al. for pristine CeO2(100) surface (0.87 eV).25” Table S48 has been corrected in the Supporting Information file and replaced by Table S48 here. The corrections mentioned do not affect the other discussions or our general conclusions. We regret any inconveniences. The authors thank Verónica Ganduglia–Pirovano and Breno L. Galvão for their valuable scientific discussions.
对 "CH4 在 (CeO2)10 簇上脱氢的 Ab Initio 研究 "的更正
遗憾的是,我们在论文中发现了两个与统一键指数-二次指数势(UBI-QEP)分析误用有关的错误,通过与 Verónica Ganduglia-Pirovano 博士和 Breno L. Galvão 博士(私人通信,2024 年 3 月 15 日)的讨论,这两个错误变得显而易见。第一个错误涉及图 5 的设计结果,我们使用 UBI-QEP 近似方法获得了活化能值。在这一分析中,我们考虑了文献中报道的 CeO2(111)表面最常见的吸附位点,而不是铈簇上最稳定的吸附位点。因此,没有保持 CH4 吸附位点与相应的 CH3 和/或 H 位点之间的依赖关系。对于我们的目的来说,更合适的方法是系统地研究 (CeO2)10 上的共吸附位点,以特定的吸附位点为参考,绘制最稳定的 CH3 或 H 位点图。因此,我们采用了上述方法,绘制了 CH3 位点图,并将最稳定的 H 位点固定为参照物;因此,第 11944 页上的文字和相关图表(图 5)应根据获得的新结果进行更新。第二个错误涉及图 6 的表述,连接结构的横向线条可能会让读者误认为是一条拟议的反应路径。实际上,我们给出的是通过 k-means 聚类法选出的每组代表性结构的反应能量值。关于图 5 和图 6 制作中的错误,必须做如下修改:在第 11946 页,"考虑到四种吸附构型,CH4 分子可以吸附在纳米簇上,[...]"这句话必须改为 "考虑到三种吸附构型,CH4 分子可以吸附在纳米簇上,[...]"这句话必须改为"[...]"。]",并将"[...](CeO2)10 纳米团簇上的 C-H 键断裂可能与 H 吸附位点[...]有关 "改为"[...](CeO2)10 纳米团簇上的 C-H 键断裂可能与 CH3 (H) 的共吸附位点[...]有关"。原图 5 应替换为本更正的图 5。第 11944 页,句子 "如图 5 所示,我们发现 CH4 在 (CeO2)10 上有四种不同的吸附模式,即伞状、反伞状、剪刀状和改良反伞状。66 在伞状和反伞状模式中,计算得出的 CH4 解离(CH4 → CH3 + H*)的势垒 (Ea) 表明与 CeO2(111) 表面相比,团簇反应活性更高。在这种情况下,我们得到的伞状模式和反伞状模式的 Ea 分别等于 0.54 eV(EaZPE = 0.48 eV)和 0.59 eV(EaZPE = 0.52 eV),小于 CeO2(111) 表面的报告值(1.44 eV)65 "必须替换为 "我们发现 CH4 在 (CeO2)10 团簇上有三种不同的吸附模式,即伞状模式、反伞状模式和剪刀模式66"。计算得出的 CH4 解离(CH4 → CH3 + H*)的势垒 (Ea) 表明,与 CeO2(111) 表面相比,簇的反应活性更高。在这种情况下,对于图 1a、b 中提出的配置,我们得到的 Ea 分别等于 0.07 eV(EaZPE = 0.03)和 0.13 eV(EaZPE = 0.09 eV),小于 CeO2(111) 表面的报告值(1.44 eV)。65" 第 11944 页中的 "那么,该状态下伞状和反伞状模式的吸附能分别为 -3.59 和 -3.95 eV,与报告的 CeO2(111) 表面的 -3.68 eV 值一致。必须删除""。原图 6 应由本更正的图 6 代替。第 11945 页,必须删除以下段落:"修改后的反伞模式和剪刀模式的能垒(分别为 0.81 和 0.64 eV)表明,与伞模式和反伞模式相比,能垒较高,这可能与 H 吸附位点有关。同样,ZPE 修正并没有使 Ea 值发生显著变化(分别为 0.74 和 0.56 eV)。修正模式和反伞模式之间的区别在于 H 的吸附位点,因为前者吸附在 3 折顶 O 位点上,而分子吸附在 2 折顶 O 位点上。而后者则相反,分子吸附在配位度更高的吸附 O 位点上。不过,改性反伞和剪刀模式得到的活化能可与 Righi 等人报告的原始 CeO2(100) 表面的活化能(0.87 eV)进行比较25。上述更正并不影响其他讨论或我们的一般性结论。如有任何不便,我们深表歉意。作者感谢 Verónica Ganduglia-Pirovano 和 Breno L. Galvão 的宝贵科学讨论。图 5.CH4 → CH3 + H 步骤中 CH4 脱氢的相对反应能量 (ΔEr)。 (a)和(b)框代表两个最稳定的(CH3 + H)/(CeO2)10 结构,它们是通过对 CH3 吸附位点扫描得到的,与最稳定的 H 样品吸附位点相对应。图 6.通过 k-means 算法选出的 (CHn + (4 - n)H)/(CeO2)10 代表性结构的相对反应能量 (ΔEr),旨在分析脱氢过程中形成新化合物的可能性。CH3 和 H 的吸附能分别为 EadCH3 和 EadH;反应焓为 ΔHr;活化能势为 Ea;相对反应能为 ΔEr。括号中的数值表示 ZPE 修正值。本文尚未被其他出版物引用。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


