High-Throughput Screening of Dual-Atom Catalysts for Methane Combustion: A Combined Density Functional Theory and Machine-Learning Study
IF 18.5
1区 材料科学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
Ceria-supported precious metal catalysts have undergone extensive investigation for the catalytic methane combustion. However, it remains a significant challenge to achieve both highly synergistic oxidation activity and efficient atom utilization remains a challenge for commonly used supported nanoparticles and single-atom catalysts. Dual-atom catalysts (DACs) emerges as a frontier of advanced catalysts, presenting unique catalytic properties that benefit from the synergy of neighboring metal sites. In this study, 361 ceria-supported DACs (M1M2/CeO2) encompassing combinations of 19 transition metals are systematically explored. Using high-throughput density functional theory calculations, the structures, stability as well as activity of M1M2/CeO2 are assessed. Notably, Au1Ga1/CeO2 is identified as a promising DAC exhibiting high activity for methane total oxidation, substantiated by comprehensive DFT-calculated reaction pathways. Furthermore, employing six machine-learning algorithms, the structure-properties relationship is explored within ceria-based DACs and highlight the importance of oxidation states and atomic radii of doped metals as the descriptors. The trained model by computational dataset exhibits high accuracy and predict a more active Mn1Au1/CeO2 than those screened using only DFT datasets. The high-throughput strategy demonstrated in this work not only provides insights into the rational design of methane oxidation catalysts, but also paves the way for exploring DACs for diverse applications.
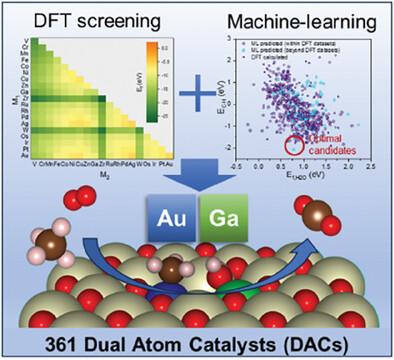
高通量筛选用于甲烷燃烧的双原子催化剂:密度泛函理论与机器学习的结合研究
在催化甲烷燃烧方面,对铈支撑贵金属催化剂进行了广泛的研究。然而,对于常用的支撑纳米颗粒和单原子催化剂来说,如何同时实现高度协同氧化活性和高效原子利用仍然是一项重大挑战。双原子催化剂(DACs)作为先进催化剂的前沿技术出现了,其独特的催化特性得益于相邻金属位点的协同作用。本研究系统地探讨了 361 种铈支撑的双原子催化剂(M1M2/CeO2),包括 19 种过渡金属的组合。通过高通量密度泛函理论计算,对 M1M2/CeO2 的结构、稳定性和活性进行了评估。值得注意的是,Au1Ga1/CeO2 被确定为一种有前途的 DAC,在甲烷全氧化方面表现出很高的活性,这一点通过全面的 DFT 计算反应路径得到了证实。此外,利用六种机器学习算法探索了铈基 DAC 的结构-性质关系,并突出了氧化态和掺杂金属原子半径作为描述因子的重要性。与仅使用 DFT 数据集筛选出的模型相比,通过计算数据集训练出的模型具有很高的准确性,并能预测出活性更强的 Mn1Au1/CeO2。这项工作中展示的高通量策略不仅为甲烷氧化催化剂的合理设计提供了启示,还为探索 DACs 的多样化应用铺平了道路。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Advanced Functional Materials
工程技术-材料科学:综合
CiteScore
29.50
自引率
4.20%
发文量
2086
审稿时长
2.1 months
期刊介绍:
Firmly established as a top-tier materials science journal, Advanced Functional Materials reports breakthrough research in all aspects of materials science, including nanotechnology, chemistry, physics, and biology every week.
Advanced Functional Materials is known for its rapid and fair peer review, quality content, and high impact, making it the first choice of the international materials science community.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


