Metal single-atom interaction with graphitic C3N4 surface based on density functional theory calculations and linear regression analysis
IF 2.4
3区 化学
Q4 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
This paper discusses the adsorption of metal single-atoms (SAs) on graphitic C3N4 (gCN) multilayered surface models based on density functional theory (DFT) calculations. We systematically searched stable adsorption sites on gCN and showed that the cavity site is preferred for metal SAs. We also discussed the usefulness of several descriptors, such as the distance between the SA and gCN, charge donation (electron transfer), and atomic radius, for evaluating the SA adsorption energy, based on the linear regression method. In addition, the most stable adsorption energy for fifth- and sixth-row metals can be predicted using only the “group” descriptor of the periodic table. The group descriptor is closely related to the atomic radius, and hence the stability of fifth- and sixth-row metals is determined from the fit between the SA radius and the gCN cavity size. The study findings facilitate the development of various metal-dopant techniques for gCN-based applications.
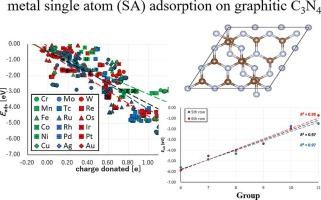
基于密度泛函理论计算和线性回归分析的金属单原子与石墨化 C3N4 表面的相互作用
本文基于密度泛函理论(DFT)计算,讨论了金属单原子(SA)在石墨化 C3N4(gCN)多层表面模型上的吸附。我们系统地搜索了 gCN 上的稳定吸附位点,结果表明空腔位点是金属 SA 的首选吸附位点。我们还基于线性回归方法,讨论了 SA 与 gCN 之间的距离、电荷捐赠(电子转移)和原子半径等几个描述因子对评估 SA 吸附能的作用。此外,仅使用元素周期表中的 "基团 "描述符就能预测第五排和第六排金属最稳定的吸附能。基团描述符与原子半径密切相关,因此第五排和第六排金属的稳定性是通过 SA 半径与 gCN 空腔尺寸之间的拟合来确定的。研究结果有助于开发各种基于 gCN 应用的金属掺杂技术。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Chemical Physics
化学-物理:原子、分子和化学物理
CiteScore
4.60
自引率
4.30%
发文量
278
审稿时长
39 days
期刊介绍:
Chemical Physics publishes experimental and theoretical papers on all aspects of chemical physics. In this journal, experiments are related to theory, and in turn theoretical papers are related to present or future experiments. Subjects covered include: spectroscopy and molecular structure, interacting systems, relaxation phenomena, biological systems, materials, fundamental problems in molecular reactivity, molecular quantum theory and statistical mechanics. Computational chemistry studies of routine character are not appropriate for this journal.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


