Design, synthesis, biological evaluation and molecular docking study of thiadiazole-isatin hybrid analogues as potential anti-diabetic and anti-bacterial agents
IF 2.5
Q2 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
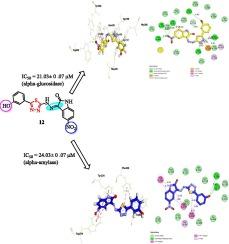
We have synthesized thiadiazole-isatin hybrid analogues (1–20), characterized through different spectroscopic techniques such as 1HNMR, 13CNMR, HREI-MS, and evaluated against α-glucosidase and α-amylase enzymes. All analogues showed potent inhibitory potentials, having an IC50 values ranged from 22.08 ± 0.09 to 54.03 ± 0.07 μM (against α-amylase) and 19.03 ± 0.04 to 50.03 ± 0.02 μM (α-glucosidase) when compared with the standard drug acarbose (IC50 = 22.07 ± 0.02 μM for α-amylase and IC50 = 18.01 ± 0.06 μM for α-glucosidase respectively). Among the series, analogues 13 (IC50 = 19.03 ± 0.04 and 22.08 ± 0.09 μM), 12 (IC50 = 21.03 ± 0.07 and 24.03 ± 0.07 μM), and 5 (IC50 = 22.02 ± 0.03 and 26.02 ± 0.04 μM) were identified as the most active inhibitors. The structure–activity relationship was carried out, mainly associated with the nature, number, position, and electron-donating and electron-withdrawing effects of the substituent (s) on the phenyl ring. With the help of molecular docking, the binding interaction of the most active analogues within the active site of enzymes was confirmed. Almost twelve compounds were also screened for antibacterial potential, and few were found to be effective against Bacillus subtilis. All compounds were verified for cytotoxicity against the 3 T3 mouse fibroblast cell line and detected non-toxic.
作为潜在抗糖尿病和抗菌剂的噻二唑-靛红杂化类似物的设计、合成、生物学评价和分子对接研究
我们合成了噻二唑-靛红杂化类似物(1-20),通过 1HNMR、13CNMR、HREI-MS 等不同光谱技术对其进行了表征,并针对α-葡萄糖苷酶和α-淀粉酶进行了评估。所有类似物都显示出强大的抑制潜力,其 IC50 值介于 22.08 ± 0.09 至 54.03 ± 0.07 μM 之间(针对α-淀粉酶)和 19.03 ± 0.04 至 50.03 ± 0.07 μM 之间(针对α-淀粉酶)。02 μM(α-葡萄糖苷酶),而标准药物阿卡波糖的 IC50 = 22.07 ± 0.02 μM(α-淀粉酶)和 IC50 = 18.01 ± 0.06 μM(α-葡萄糖苷酶)。在这一系列中,类似物 13(IC50 = 19.03 ± 0.04 和 22.08 ± 0.09 μM)、12(IC50 = 21.03 ± 0.07 和 24.03 ± 0.07 μM)和 5(IC50 = 22.02 ± 0.03 和 26.02 ± 0.04 μM)被确定为最有效的抑制剂。结构与活性的关系主要与苯环上取代基的性质、数目、位置、电子供取效应有关。在分子对接的帮助下,确认了最具活性的类似物在酶活性位点内的结合相互作用。还对近 12 种化合物进行了抗菌潜力筛选,发现其中少数化合物对枯草杆菌有效。所有化合物都对 3 T3 小鼠成纤维细胞系进行了细胞毒性验证,并检测出无毒性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


