Suliman Adam, Itamar Kass, Dana Krepel-Zussman, Gal Masarati, Dorit Shemesh and Avital Sharir-Ivry*,
{"title":"Effect of Protein-Polarized Ligand Charges on Relative Protein Ligand Binding Affinities","authors":"Suliman Adam, Itamar Kass, Dana Krepel-Zussman, Gal Masarati, Dorit Shemesh and Avital Sharir-Ivry*, ","doi":"10.1021/acs.jctc.3c0133710.1021/acs.jctc.3c01337","DOIUrl":null,"url":null,"abstract":"<p >A major challenge in computer-aided drug design is predicting relative binding energies of different molecules to a target protein using fast and accurate free-energy calculation methods. Free-energy calculations are primarily computed by utilizing classical molecular dynamics simulations based on all-atom force fields (FF) to model the interactions in the system. The present standard classical all-atom FFs contain fixed partial charges on the atoms, and hence electrostatic interactions are modeled between them. The parametrization process to determine these partial charges usually relies on quantum mechanics or semiempirical calculations of the molecule in the gas phase or homogeneous water surrounding. These present standard parametrization schemes of the partial charges neglect, therefore, polarization effects from the protein surrounding. The absence of protein polarization effects can lead to significant errors in free-energy calculations in proteins. We present a parametrization scheme for the partial charges of ligands, named protein-induced polarization (PIP) charges, which account for the electrostatic polarization due to the protein surrounding. The scheme involves single-point quantum mechanics/molecular mechanics calculations of the ligand charges in the protein/water surrounding. Using PIP ligand partial charges, we have calculated the relative binding free energies (RBFEs) of well-studied protein−ligand systems. We show here that RBFEs computed with PIP charges are either significantly improved or at least comparable to those computed with nonpolarized standard GAFF charges. Overall, we present a simple-to-use parametrization scheme to include protein polarization in any type of binding free-energy calculations. The parametrization scheme increases the accuracy in RBFE calculations, while it does not add significant computation time to standard parametrization procedures.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":null,"pages":null},"PeriodicalIF":5.7000,"publicationDate":"2024-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.3c01337","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
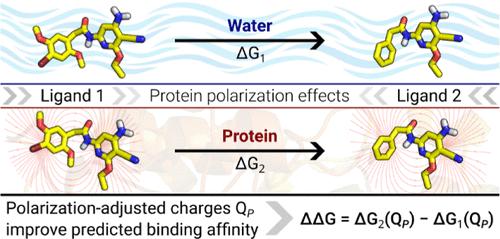
A major challenge in computer-aided drug design is predicting relative binding energies of different molecules to a target protein using fast and accurate free-energy calculation methods. Free-energy calculations are primarily computed by utilizing classical molecular dynamics simulations based on all-atom force fields (FF) to model the interactions in the system. The present standard classical all-atom FFs contain fixed partial charges on the atoms, and hence electrostatic interactions are modeled between them. The parametrization process to determine these partial charges usually relies on quantum mechanics or semiempirical calculations of the molecule in the gas phase or homogeneous water surrounding. These present standard parametrization schemes of the partial charges neglect, therefore, polarization effects from the protein surrounding. The absence of protein polarization effects can lead to significant errors in free-energy calculations in proteins. We present a parametrization scheme for the partial charges of ligands, named protein-induced polarization (PIP) charges, which account for the electrostatic polarization due to the protein surrounding. The scheme involves single-point quantum mechanics/molecular mechanics calculations of the ligand charges in the protein/water surrounding. Using PIP ligand partial charges, we have calculated the relative binding free energies (RBFEs) of well-studied protein−ligand systems. We show here that RBFEs computed with PIP charges are either significantly improved or at least comparable to those computed with nonpolarized standard GAFF charges. Overall, we present a simple-to-use parametrization scheme to include protein polarization in any type of binding free-energy calculations. The parametrization scheme increases the accuracy in RBFE calculations, while it does not add significant computation time to standard parametrization procedures.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


