Bilgesu Ak, Erhan Parıltay, Reyhan Gümüşburun, Ceyda Tunakan Dalgıç, Ayça Aykut, Asude Durmaz, Haluk Akın, Ömür Ardeniz, Bernice Lo
{"title":"Uniparental Disomy of Chromosome 4: A Case of Whole Chromosome UPD Presenting with LRBA Deficiency","authors":"Bilgesu Ak, Erhan Parıltay, Reyhan Gümüşburun, Ceyda Tunakan Dalgıç, Ayça Aykut, Asude Durmaz, Haluk Akın, Ömür Ardeniz, Bernice Lo","doi":"10.1007/s10875-024-01803-9","DOIUrl":null,"url":null,"abstract":"<h3 data-test=\"abstract-sub-heading\">Purpose</h3><p>Lipopolysaccharide-responsive beige-like anchor protein (<i>LRBA</i>) encodes a widely expressed cytosolic protein that participates in polarized vesicle trafficking. Homozygous loss-of-function <i>LRBA</i> mutations can lead to immune deficiency due to the lack of immune regulation, classified as a part of Tregopathies. We present a case of a 49-year-old female, with polyarthralgia in metacarpophalangeal and proximal interphalangeal joints bilaterally, and morning stiffness, leading to the diagnosis of rheumatoid arthritis treated with pulse steroid therapy. She had experienced sepsis and in-depth scrutiny revealed panhypogammaglobulinemia. After being referred to the immunology clinic, she was followed under the diagnosis of common variable immunodeficiency (CVID)-like inborn errors of immunity (IEI).</p><h3 data-test=\"abstract-sub-heading\">Methods and Results</h3><p>Physical examination and diagnostic follow-up revealed massive splenomegaly accompanied by portal hypertension, and ulcerations in the colon. She also presented with periodic hematuria and dysuria. Cystoscopic biopsy revealed mast cell-derived interstitial cystitis which has not been previously reported in LRBA deficiency in the literature to our knowledge. A multi-gene next-generation sequencing panel performed for immune deficiencies (264 genes and 524 amplicons), resulted in the identification of an apparently homozygous <i>LRBA</i> mutation (p.Arg722His) in the Beige and Chediak-Higashi (BEACH) domain. The SNP array showed copy neutral absence of heterozygosity of the entire chromosome 4, which is consistent with uniparental isodisomy of chromosome 4.</p><h3 data-test=\"abstract-sub-heading\">Conclusion</h3><p>In conclusion, this case study underscores the critical role of LRBA in immune regulation and highlights the clinical heterogeneity associated with LRBA deficiency. The patient’s presentation with severe immune dysregulation, including massive splenomegaly, portal hypertension, and the novel finding of mast cell-derived interstitial cystitis, expands the clinical spectrum of <i>LRBA</i> mutations. The identification of an apparently homozygous <i>LRBA</i> mutation via next-generation sequencing further emphasizes the importance of genetic analysis in diagnosing monogenic defects manifested as CVID-like phenotype. This is the first reported case of LRBA deficiency due to whole chromosome UPD to our knowledge. Future research should focus on elucidating the full range of clinical manifestations and developing targeted therapies for patients with LRBA deficiency.</p>","PeriodicalId":15531,"journal":{"name":"Journal of Clinical Immunology","volume":"7 1","pages":""},"PeriodicalIF":7.2000,"publicationDate":"2024-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10875-024-01803-9","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Purpose
Lipopolysaccharide-responsive beige-like anchor protein (LRBA) encodes a widely expressed cytosolic protein that participates in polarized vesicle trafficking. Homozygous loss-of-function LRBA mutations can lead to immune deficiency due to the lack of immune regulation, classified as a part of Tregopathies. We present a case of a 49-year-old female, with polyarthralgia in metacarpophalangeal and proximal interphalangeal joints bilaterally, and morning stiffness, leading to the diagnosis of rheumatoid arthritis treated with pulse steroid therapy. She had experienced sepsis and in-depth scrutiny revealed panhypogammaglobulinemia. After being referred to the immunology clinic, she was followed under the diagnosis of common variable immunodeficiency (CVID)-like inborn errors of immunity (IEI).
Methods and Results
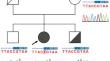
Physical examination and diagnostic follow-up revealed massive splenomegaly accompanied by portal hypertension, and ulcerations in the colon. She also presented with periodic hematuria and dysuria. Cystoscopic biopsy revealed mast cell-derived interstitial cystitis which has not been previously reported in LRBA deficiency in the literature to our knowledge. A multi-gene next-generation sequencing panel performed for immune deficiencies (264 genes and 524 amplicons), resulted in the identification of an apparently homozygous LRBA mutation (p.Arg722His) in the Beige and Chediak-Higashi (BEACH) domain. The SNP array showed copy neutral absence of heterozygosity of the entire chromosome 4, which is consistent with uniparental isodisomy of chromosome 4.
Conclusion
In conclusion, this case study underscores the critical role of LRBA in immune regulation and highlights the clinical heterogeneity associated with LRBA deficiency. The patient’s presentation with severe immune dysregulation, including massive splenomegaly, portal hypertension, and the novel finding of mast cell-derived interstitial cystitis, expands the clinical spectrum of LRBA mutations. The identification of an apparently homozygous LRBA mutation via next-generation sequencing further emphasizes the importance of genetic analysis in diagnosing monogenic defects manifested as CVID-like phenotype. This is the first reported case of LRBA deficiency due to whole chromosome UPD to our knowledge. Future research should focus on elucidating the full range of clinical manifestations and developing targeted therapies for patients with LRBA deficiency.
期刊介绍:
The Journal of Clinical Immunology publishes impactful papers in the realm of human immunology, delving into the diagnosis, pathogenesis, prognosis, or treatment of human diseases. The journal places particular emphasis on primary immunodeficiencies and related diseases, encompassing inborn errors of immunity in a broad sense, their underlying genotypes, and diverse phenotypes. These phenotypes include infection, malignancy, allergy, auto-inflammation, and autoimmunity. We welcome a broad spectrum of studies in this domain, spanning genetic discovery, clinical description, immunologic assessment, diagnostic approaches, prognosis evaluation, and treatment interventions. Case reports are considered if they are genuinely original and accompanied by a concise review of the relevant medical literature, illustrating how the novel case study advances the field. The instructions to authors provide detailed guidance on the four categories of papers accepted by the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


