{"title":"In-depth investigation of the iodine-nitrogen interaction mechanism using ab initio and density functional theory calculations","authors":"","doi":"10.1016/j.comptc.2024.114862","DOIUrl":null,"url":null,"abstract":"<div><p>In this paper, we<!--> <!-->systematically<!--> <!-->investigate the interactions of I<sub>2</sub> <!-->with several representative nitrogen-containing building units (NBUs) using high-precision<em> <!-->ab</em> <em>initio</em> <!-->and density functional theory calculations. Our findings reveal that the binding strengths of I<sub>2</sub> <!-->with NBUs are<!--> <!-->positively correlated with<!--> <!-->the hybridization degrees<!--> <!-->of N-<em>2p</em> <!-->electrons. Further energy analyses<!--> <!-->indicate that the I-N orbital interaction determines<!--> <!-->the binding strength to a great extent. Especially for I<sub>2</sub>@NBU(<em>sp<sup>3</sup></em>), its<!--> <em>σn</em> <!-->binding orbital formed by the<!--> <em>σ</em> <!-->orbital of I<sub>2</sub> <!-->and the <em>n</em> orbital of NBU(<em>sp<sup>3</sup></em>) exhibits nearly perfect orbital overlap. QTAIM analyses identify<!--> <!-->the unique partially covalent feature of I-N bonds in I<sub>2</sub>@NBU(<em>sp<sup>2</sup></em>/<em>sp<sup>3</sup></em>) and the pure ionic feature in I<sub>2</sub>@NBU(<em>sp</em>). The I-N orbital interactions lead to remarkable<!--> <!-->electron reorganizations in I<sub>2</sub>@NBUs. Moreover, we demonstrate that<!--> <!-->the binding strengths between NBUs and I<sub>2</sub> can be effectively regulated through introducing electron-donating/electron-withdrawing functional groups. This study deepens the fundamental understanding of iodine coordination chemistry.</p></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":null,"pages":null},"PeriodicalIF":3.0000,"publicationDate":"2024-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004018","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
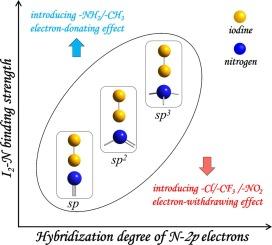
In this paper, we systematically investigate the interactions of I2 with several representative nitrogen-containing building units (NBUs) using high-precision abinitio and density functional theory calculations. Our findings reveal that the binding strengths of I2 with NBUs are positively correlated with the hybridization degrees of N-2p electrons. Further energy analyses indicate that the I-N orbital interaction determines the binding strength to a great extent. Especially for I2@NBU(sp3), its σn binding orbital formed by the σ orbital of I2 and the n orbital of NBU(sp3) exhibits nearly perfect orbital overlap. QTAIM analyses identify the unique partially covalent feature of I-N bonds in I2@NBU(sp2/sp3) and the pure ionic feature in I2@NBU(sp). The I-N orbital interactions lead to remarkable electron reorganizations in I2@NBUs. Moreover, we demonstrate that the binding strengths between NBUs and I2 can be effectively regulated through introducing electron-donating/electron-withdrawing functional groups. This study deepens the fundamental understanding of iodine coordination chemistry.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


