Molecular geometry specific Monte Carlo simulation of the efficacy of diamond crystal formation from diamondoids
IF 5.9
2区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
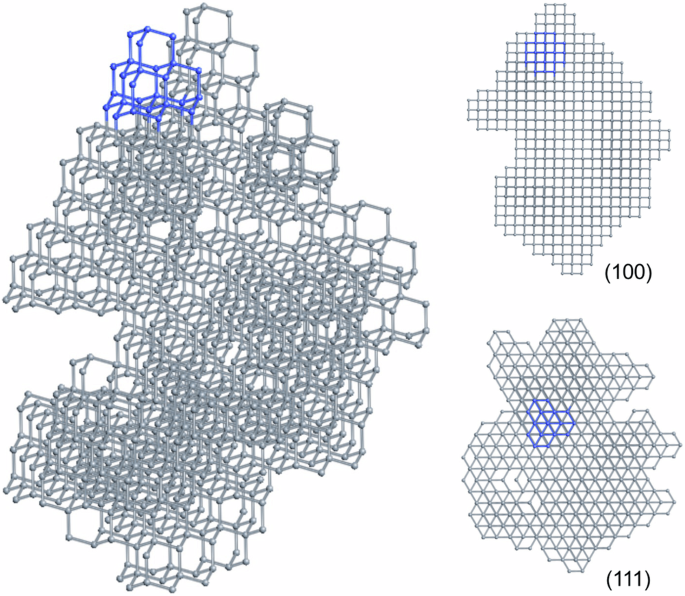
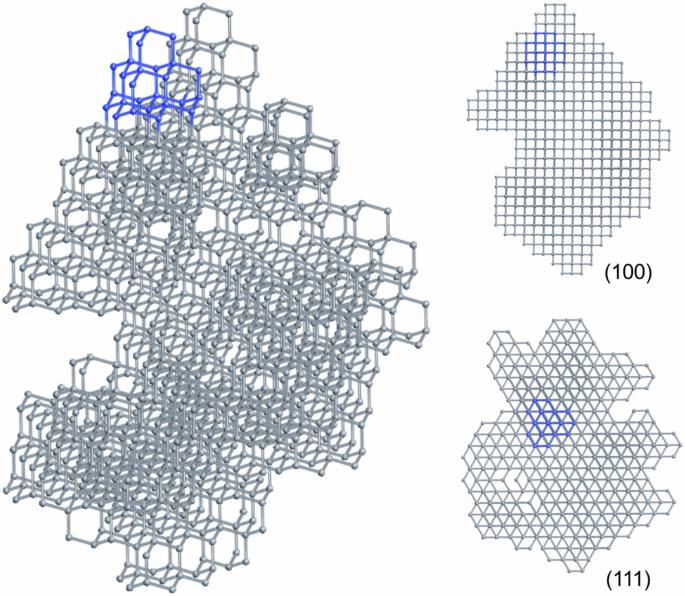
Diamondoids are a class of organic molecules with the carbon skeletons isostructural to nano-diamond, and have been shown to be promising precursors for diamond formation. In this work, the formation of diamond crystals from various diamondoid molecule building blocks was studied using our developed molecular geometry specific Monte Carlo method. We maintained the internal carbon skeletons of the diamondoid molecules, and investigated how the carbon-carbon bonds form between diamondoid molecules and how efficient the process is to form diamond crystals. The simulations show that higher diamondoid molecules can produce structures closer to a diamond crystal compared with lower diamondoid molecules. Specifically, using higher diamondoid molecules, larger bulk diamond crystals are formed with fewer vacancies. The higher propensity of certain diamondoids to form diamond crystals reveals insights into the microscopic processes of diamond formation under high-pressure high-temperature conditions. Diamondoids are a series of hydrogen-terminated nanometer-sized hydrocarbons that can be used to synthesize high-quality diamond crystals. Here, the authors use Monte Carlo simulations to study the potentials of different diamondoids in constructing diamond crystals with the assumption that the carbon skeletons keep intact, and find that higher diamondoid molecules are most suitable.
针对金刚石晶体形成效率的分子几何蒙特卡洛模拟
类金刚石是一类碳骨架与纳米金刚石结构相同的有机分子,已被证明是有望形成金刚石的前体。在这项研究中,我们利用所开发的分子几何特定蒙特卡洛方法,研究了各种类金刚石分子构建模块形成金刚石晶体的过程。我们保留了类金刚石分子的内部碳骨架,并研究了类金刚石分子之间如何形成碳碳键以及形成金刚石晶体的效率如何。模拟结果表明,与较低的类金刚石分子相比,较高的类金刚石分子能产生更接近金刚石晶体的结构。具体来说,使用较高的类金刚石分子可以形成较大的块状金刚石晶体,空位较少。某些类金刚石更倾向于形成金刚石晶体,这揭示了在高压高温条件下形成金刚石的微观过程。类金刚石是一系列氢端纳米级碳氢化合物,可用于合成高质量的金刚石晶体。在此,作者利用蒙特卡洛模拟研究了不同类金刚石在构建金刚石晶体时的潜力,并假设碳骨架保持完好无损,结果发现高类金刚石分子最适合构建金刚石晶体。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊
Communications Chemistry
Chemistry-General Chemistry
CiteScore
7.70
自引率
1.70%
发文量
146
审稿时长
13 weeks
期刊介绍:
Communications Chemistry is an open access journal from Nature Research publishing high-quality research, reviews and commentary in all areas of the chemical sciences. Research papers published by the journal represent significant advances bringing new chemical insight to a specialized area of research. We also aim to provide a community forum for issues of importance to all chemists, regardless of sub-discipline.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


