{"title":"From Challenge to Opportunity: How Shwachman–Diamond Syndrome Became a Promising Target for Therapy Development","authors":"Eszter S. Hars, Lisa J. McReynolds","doi":"10.1002/cpt.3393","DOIUrl":null,"url":null,"abstract":"<p>\n <b>Rare diseases affect over 30 million people in the United States and 300 million globally, yet 95% lack FDA-approved treatments. Rare disease therapy development poses unique challenges and opportunities. Shwachman–Diamond syndrome (SDS) is emerging as a model rare disease due to its uniform genetics, robust molecular understanding, and mature research infrastructure—including de-risking through model development, regulatory engagement, patient community development, ICD-10 implementation, and data access, driven by the SDS Alliance—a research-focused patient advocacy organization.</b>\n </p><p>Rare disease connects us all. Every one of us knows someone impacted by one of over 10,000 rare diseases, which combined affect 300 million people globally and over 30 million people in the US—nearly 1 in 10. 95% of rare diseases have no FDA-approved treatments, but the tides are changing. Drugs that target rare diseases represent over half of all novel drugs and biologics approved by the FDA in recent years. This did not happen overnight or by chance. The Orphan Drug Act of 1983 and the relentless advocacy of leading rare disease organizations made this possible.<span><sup>1</sup></span></p><p>Developing drugs for rare diseases poses unique challenges and opportunities. Beyond financial considerations, developers face hurdles such as recruiting enough patients for clinical trials, obtaining natural history data, and designing clinical trials that can convincingly demonstrate safety and efficacy to regulators and patients. Opportunities include new niche markets with substantial financial potential and the intangible rewards of bringing therapies to patients in desperate need, witnessing firsthand the life-changing positive impact on families and communities.</p><p>Given the large number of distinct rare diseases and the high risks involved in therapy development, what makes a rare disease a compelling target for investment? The more that is known about the disease mechanism, the more that patients are connected, and the more that relevant research tools and infrastructure are developed, the better. But often it takes serendipity and time. Small molecule drugs developed for a common condition may be a great fit for a rare disease (drug repurposing) based on its mechanism of action. Modern gene editing tools open doors for addressing the underlying cause of rare genetic disorders. Precision medicine is the new frontier, offering hope not only to rare disease patients but to all of us.</p><p>SDS is a rare genetic disorder that affects about 1:100,000 births, and thousands of people globally, many undiagnosed due to variable presentation of symptoms.<span><sup>2, 3</sup></span> It impacts multiple organ systems and significantly increases the risk of leukemia, with poor outcomes.<span><sup>4, 5</sup></span> By age 30, about 30% of SDS patients develop acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), or severe bone marrow failure, often necessitating a hematopoietic cell transplant (HCT)—a high-risk procedure with uncertain long-term effects. The outcomes for AML in SDS patients are particularly grim.<span><sup>4</sup></span></p><p>Other common symptoms include failure to thrive, pain, and malnutrition due to exocrine pancreatic insufficiency (EPI), elevated liver enzymes, respiratory issues, skeletal abnormalities, and cognitive delays.<span><sup>2, 3</sup></span> The severity of symptoms varies widely from patient to patient, causing frequently missed or delayed diagnoses, and putting patients at risk for unnecessary suffering and life-threatening complications.</p><p>Most cases of SDS are caused by pathogenic variants in the essential gene <i>SBDS</i>,<span><sup>6</sup></span> typically inherited from unsuspecting carrier parents in an autosomal recessive pattern, or caused <i>de-novo</i> by gene conversion/recombination with an adjacent pseudogene, <i>SBDSP1</i>. The SBDS protein plays a critical role in ribosome biogenesis by catalyzing the displacement of eIF6 from the large ribosomal subunit, thereby allowing the large and small ribosomal subunits to join and form a working ribosome.<span><sup>6</sup></span> Ribosomes are large protein complexes that translate the genetic information encoded in mRNA into proteins, playing a key role in the central dogma of molecular biology and being fundamental to life. In SDS, there are not enough working ribosomes to meet the protein demand of cells. Recent publications suggest that the highly increased risk of developing leukemia is due to “reduced fitness” of hematopoietic stem cells (HSCs), causing a selection pressure favoring cells that acquire maladaptive somatic mutations, such as TP53.<span><sup>6, 7</sup></span></p><p>Currently, there is no targeted therapy for SDS. Patients are left anxious about possible life-threatening complications and invasive surveillance to look for concerning findings to trigger HCT. On the positive side, day-to-day quality of life has significantly improved thanks to advances in symptomatic treatments, such as pancreatic enzyme replacement therapy (PERT) to manage the effects of EPI, granulocyte-colony stimulating factor (G-CSF) to improve neutropenia, and others.</p><p>There is a wide range of pre-clinical research efforts aimed at addressing the underlying cause of SDS. Several programs are exploring base- and prime editing to fix the genetic cause; there are small molecule screening and development efforts to de-stabilize the binding of eIF6 to overcome the SBDS defect, based on learning from somatic genetic rescue<span><sup>8</sup></span>; antisense oligonucleotides (ASOs) have been shown to increase wild-type SBDS expression; plus, additional programs are looking at targeting other common SBDS pathogenic variants with nonsense suppressor drugs.<span><sup>9</sup></span></p><p>SDS has several characteristics that make it an ideal model rare disease and target for therapy development (see <b>Figure</b> 1).</p><p>First, the genetics are well understood and extremely uniform. Over 90% of patients not only have pathogenic variants in the same gene (<i>SBDS</i>) but also have at least one copy of a specific splice-site mutation (known as c.258+2T>C). This common variant allows a small amount of wild-type protein to be made, making SDS compatible with life, and making it a good candidate for several therapeutic modalities.<span><sup>10</sup></span></p><p>Second, the primary organ system of concern, the bone marrow and in particular the HSCs, are readily accessible by a variety of therapeutic modalities, including small molecules and gene therapy (i.e., base and prime editing)—both <i>ex vivo</i> (via HCT as in severe combined immunodeficiency (SCID) and sickle cell disease) and <i>in vivo</i> (a rapidly evolving field).</p><p>Third, due to the long history of SDS research, several natural history studies have been collecting clinical data for years or decades and are ready to support clinical development. Additional data resources are available as well, as discussed below.</p><p>To further de-risk SDS therapy development, the Shwachman-Diamond Syndrome Alliance—a global research-focused patient advocacy nonprofit organization—has launched several programs to develop critical research tools and infrastructure. These programs fall into four broad categories: model systems, regulatory engagement, patient community development, and data access (see <b>Figure</b> 1).</p><p>Reflecting on the SDS Alliance's experience with positioning SDS as a model rare disease and emulating the strategies of successful rare disease organizations in recent years, it is evident that modest investments in strategic tools and infrastructure can significantly de-risk rare diseases and incentivize new therapeutic development and drug repurposing. We hope that other rare disease communities will benefit from these approaches and that drug developers continue to recognize the value of partnering with patient communities to pursue therapies and cures for the thousands of rare diseases affecting millions of people. Shwachman–Diamond syndrome is ready.</p><p>This work was supported by The Chan Zuckerberg Initiative, a research grant from the University of Pennsylvania Orphan Disease Center in partnership with the Shwachman-Diamond Syndrome Alliance, and the SDS patient community. LJM was funded by the Intramural Research Program of the National Cancer Institute.</p><p>The authors declare no competing interests for this work.</p>","PeriodicalId":153,"journal":{"name":"Clinical Pharmacology & Therapeutics","volume":"116 6","pages":"1377-1380"},"PeriodicalIF":6.3000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11567794/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pharmacology & Therapeutics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cpt.3393","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
Rare diseases affect over 30 million people in the United States and 300 million globally, yet 95% lack FDA-approved treatments. Rare disease therapy development poses unique challenges and opportunities. Shwachman–Diamond syndrome (SDS) is emerging as a model rare disease due to its uniform genetics, robust molecular understanding, and mature research infrastructure—including de-risking through model development, regulatory engagement, patient community development, ICD-10 implementation, and data access, driven by the SDS Alliance—a research-focused patient advocacy organization.
Rare disease connects us all. Every one of us knows someone impacted by one of over 10,000 rare diseases, which combined affect 300 million people globally and over 30 million people in the US—nearly 1 in 10. 95% of rare diseases have no FDA-approved treatments, but the tides are changing. Drugs that target rare diseases represent over half of all novel drugs and biologics approved by the FDA in recent years. This did not happen overnight or by chance. The Orphan Drug Act of 1983 and the relentless advocacy of leading rare disease organizations made this possible.1
Developing drugs for rare diseases poses unique challenges and opportunities. Beyond financial considerations, developers face hurdles such as recruiting enough patients for clinical trials, obtaining natural history data, and designing clinical trials that can convincingly demonstrate safety and efficacy to regulators and patients. Opportunities include new niche markets with substantial financial potential and the intangible rewards of bringing therapies to patients in desperate need, witnessing firsthand the life-changing positive impact on families and communities.
Given the large number of distinct rare diseases and the high risks involved in therapy development, what makes a rare disease a compelling target for investment? The more that is known about the disease mechanism, the more that patients are connected, and the more that relevant research tools and infrastructure are developed, the better. But often it takes serendipity and time. Small molecule drugs developed for a common condition may be a great fit for a rare disease (drug repurposing) based on its mechanism of action. Modern gene editing tools open doors for addressing the underlying cause of rare genetic disorders. Precision medicine is the new frontier, offering hope not only to rare disease patients but to all of us.
SDS is a rare genetic disorder that affects about 1:100,000 births, and thousands of people globally, many undiagnosed due to variable presentation of symptoms.2, 3 It impacts multiple organ systems and significantly increases the risk of leukemia, with poor outcomes.4, 5 By age 30, about 30% of SDS patients develop acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), or severe bone marrow failure, often necessitating a hematopoietic cell transplant (HCT)—a high-risk procedure with uncertain long-term effects. The outcomes for AML in SDS patients are particularly grim.4
Other common symptoms include failure to thrive, pain, and malnutrition due to exocrine pancreatic insufficiency (EPI), elevated liver enzymes, respiratory issues, skeletal abnormalities, and cognitive delays.2, 3 The severity of symptoms varies widely from patient to patient, causing frequently missed or delayed diagnoses, and putting patients at risk for unnecessary suffering and life-threatening complications.
Most cases of SDS are caused by pathogenic variants in the essential gene SBDS,6 typically inherited from unsuspecting carrier parents in an autosomal recessive pattern, or caused de-novo by gene conversion/recombination with an adjacent pseudogene, SBDSP1. The SBDS protein plays a critical role in ribosome biogenesis by catalyzing the displacement of eIF6 from the large ribosomal subunit, thereby allowing the large and small ribosomal subunits to join and form a working ribosome.6 Ribosomes are large protein complexes that translate the genetic information encoded in mRNA into proteins, playing a key role in the central dogma of molecular biology and being fundamental to life. In SDS, there are not enough working ribosomes to meet the protein demand of cells. Recent publications suggest that the highly increased risk of developing leukemia is due to “reduced fitness” of hematopoietic stem cells (HSCs), causing a selection pressure favoring cells that acquire maladaptive somatic mutations, such as TP53.6, 7
Currently, there is no targeted therapy for SDS. Patients are left anxious about possible life-threatening complications and invasive surveillance to look for concerning findings to trigger HCT. On the positive side, day-to-day quality of life has significantly improved thanks to advances in symptomatic treatments, such as pancreatic enzyme replacement therapy (PERT) to manage the effects of EPI, granulocyte-colony stimulating factor (G-CSF) to improve neutropenia, and others.
There is a wide range of pre-clinical research efforts aimed at addressing the underlying cause of SDS. Several programs are exploring base- and prime editing to fix the genetic cause; there are small molecule screening and development efforts to de-stabilize the binding of eIF6 to overcome the SBDS defect, based on learning from somatic genetic rescue8; antisense oligonucleotides (ASOs) have been shown to increase wild-type SBDS expression; plus, additional programs are looking at targeting other common SBDS pathogenic variants with nonsense suppressor drugs.9
SDS has several characteristics that make it an ideal model rare disease and target for therapy development (see Figure 1).
First, the genetics are well understood and extremely uniform. Over 90% of patients not only have pathogenic variants in the same gene (SBDS) but also have at least one copy of a specific splice-site mutation (known as c.258+2T>C). This common variant allows a small amount of wild-type protein to be made, making SDS compatible with life, and making it a good candidate for several therapeutic modalities.10
Second, the primary organ system of concern, the bone marrow and in particular the HSCs, are readily accessible by a variety of therapeutic modalities, including small molecules and gene therapy (i.e., base and prime editing)—both ex vivo (via HCT as in severe combined immunodeficiency (SCID) and sickle cell disease) and in vivo (a rapidly evolving field).
Third, due to the long history of SDS research, several natural history studies have been collecting clinical data for years or decades and are ready to support clinical development. Additional data resources are available as well, as discussed below.
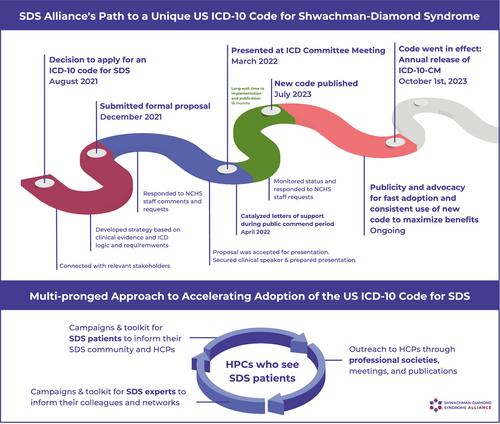
To further de-risk SDS therapy development, the Shwachman-Diamond Syndrome Alliance—a global research-focused patient advocacy nonprofit organization—has launched several programs to develop critical research tools and infrastructure. These programs fall into four broad categories: model systems, regulatory engagement, patient community development, and data access (see Figure 1).
Reflecting on the SDS Alliance's experience with positioning SDS as a model rare disease and emulating the strategies of successful rare disease organizations in recent years, it is evident that modest investments in strategic tools and infrastructure can significantly de-risk rare diseases and incentivize new therapeutic development and drug repurposing. We hope that other rare disease communities will benefit from these approaches and that drug developers continue to recognize the value of partnering with patient communities to pursue therapies and cures for the thousands of rare diseases affecting millions of people. Shwachman–Diamond syndrome is ready.
This work was supported by The Chan Zuckerberg Initiative, a research grant from the University of Pennsylvania Orphan Disease Center in partnership with the Shwachman-Diamond Syndrome Alliance, and the SDS patient community. LJM was funded by the Intramural Research Program of the National Cancer Institute.
The authors declare no competing interests for this work.
期刊介绍:
Clinical Pharmacology & Therapeutics (CPT) is the authoritative cross-disciplinary journal in experimental and clinical medicine devoted to publishing advances in the nature, action, efficacy, and evaluation of therapeutics. CPT welcomes original Articles in the emerging areas of translational, predictive and personalized medicine; new therapeutic modalities including gene and cell therapies; pharmacogenomics, proteomics and metabolomics; bioinformation and applied systems biology complementing areas of pharmacokinetics and pharmacodynamics, human investigation and clinical trials, pharmacovigilence, pharmacoepidemiology, pharmacometrics, and population pharmacology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


