Abolfazl Olyaei, Monir Shalbafan and Mahdieh Sadeghpour
{"title":"Bioactivity of benzophenazine enaminone derivatives as inhibitors of ERK2 based on molecular docking and dynamics simulation studies","authors":"Abolfazl Olyaei, Monir Shalbafan and Mahdieh Sadeghpour","doi":"10.1039/D4NJ00983E","DOIUrl":null,"url":null,"abstract":"<p >Molecular docking is a commonly employed technique in structure-based drug design that generates the binding pose and affinity between ligands and targets. In the present study, nine benzo[<em>a</em>]phenazin enaminone derivatives containing substituted aryl and heteroaryl amines were subjected to molecular docking (Auto Dock 4.2) studies for the inhibition of ERK2 (extracellular signal-regulated kinase 2). The <em>in silico</em> molecular docking study results showed that compounds <strong>D</strong> and <strong>E</strong> have minimum binding energy and have good affinity toward the active pocket. The compounds demonstrated comparable binding free energies, each at −10.5 kcal mol<small><sup>−1</sup></small>, with the ligand binding sites located at ASP109(A) and the 1-H bonds positioned within a distance range of 2.98–2.99 Å. However, these compounds showed a higher ERK2 inhibitory effect compared to purvalanol, the standard drug, which had a binding free energy of −7.5 kcal mol<small><sup>−1</sup></small>. Purvalanol bound to the ligand binding sites at Met106(A) and Asn152(A), with the 2-H bonds situated at distances of 2.99 Å and 3.17 Å, respectively. Furthermore, the RMSD value for compounds <strong>D</strong> and <strong>E</strong> stabilizes around a consistent fixed value of 1.5, indicating a stable protein conformation. Moreover, molecular dynamics (MD) studies were conducted to evaluate the stability of the docked complexes with ligands <strong>D</strong> and <strong>E</strong>. We hypothesize that these compounds may inhibit ERK2 and could potentially be used as drugs for cancer treatment. Therefore, they can be regarded as potent inhibitors of ERK2 and effective anticancer compounds for therapeutic application.</p>","PeriodicalId":95,"journal":{"name":"New Journal of Chemistry","volume":null,"pages":null},"PeriodicalIF":2.7000,"publicationDate":"2024-06-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"New Journal of Chemistry","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/nj/d4nj00983e","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
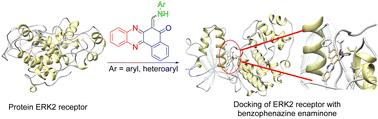
Molecular docking is a commonly employed technique in structure-based drug design that generates the binding pose and affinity between ligands and targets. In the present study, nine benzo[a]phenazin enaminone derivatives containing substituted aryl and heteroaryl amines were subjected to molecular docking (Auto Dock 4.2) studies for the inhibition of ERK2 (extracellular signal-regulated kinase 2). The in silico molecular docking study results showed that compounds D and E have minimum binding energy and have good affinity toward the active pocket. The compounds demonstrated comparable binding free energies, each at −10.5 kcal mol−1, with the ligand binding sites located at ASP109(A) and the 1-H bonds positioned within a distance range of 2.98–2.99 Å. However, these compounds showed a higher ERK2 inhibitory effect compared to purvalanol, the standard drug, which had a binding free energy of −7.5 kcal mol−1. Purvalanol bound to the ligand binding sites at Met106(A) and Asn152(A), with the 2-H bonds situated at distances of 2.99 Å and 3.17 Å, respectively. Furthermore, the RMSD value for compounds D and E stabilizes around a consistent fixed value of 1.5, indicating a stable protein conformation. Moreover, molecular dynamics (MD) studies were conducted to evaluate the stability of the docked complexes with ligands D and E. We hypothesize that these compounds may inhibit ERK2 and could potentially be used as drugs for cancer treatment. Therefore, they can be regarded as potent inhibitors of ERK2 and effective anticancer compounds for therapeutic application.
分子对接是基于结构的药物设计中常用的一种技术,可生成配体与靶点之间的结合姿态和亲和力。本研究对含有取代芳基和杂芳基胺的九种苯并[a]吩嗪烯酮衍生物进行了分子对接(Auto Dock 4.2)研究,以抑制 ERK2(细胞外信号调节激酶 2)。硅学分子对接研究结果表明,化合物 D 和 E 的结合能最小,对活性口袋具有良好的亲和力。这两个化合物的结合自由能相当,均为-10.5 kcal/mol,配体结合位点位于 ASP109(A),1-H 键的距离范围为 2.98-2.99 Å。Purvalanol 与 Met106(A) 和 Asn152(A) 的配体结合位点结合,2-H 键的距离分别为 2.99 Å 和 3.17 Å。此外,化合物 D 和 E 的 RMSD 值稳定在一致的固定值 1.5 左右,表明蛋白质构象稳定。此外,我们还进行了分子动力学(MD)研究,以评估与配体 D 和 E 的对接复合物的稳定性。因此,它们可被视为 ERK2 的强效抑制剂和有效的抗癌化合物,可用于治疗。

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


