Efficient Spin-Adapted Implementation of Multireference Algebraic Diagrammatic Construction Theory. I. Core-Ionized States and X-ray Photoelectron Spectra
Carlos E. V. de Moura*, and , Alexander Yu. Sokolov*,
{"title":"Efficient Spin-Adapted Implementation of Multireference Algebraic Diagrammatic Construction Theory. I. Core-Ionized States and X-ray Photoelectron Spectra","authors":"Carlos E. V. de Moura*, and , Alexander Yu. Sokolov*, ","doi":"10.1021/acs.jpca.4c03161","DOIUrl":null,"url":null,"abstract":"<p >We present an efficient implementation of multireference algebraic diagrammatic construction theory (MR-ADC) for simulating core-ionized states and X-ray photoelectron spectra (XPS). Taking advantage of spin adaptation, automatic code generation, and density fitting, our implementation can perform calculations for molecules with more than 1500 molecular orbitals, incorporating static and dynamic correlation in the ground and excited electronic states. We demonstrate the capabilities of MR-ADC methods by simulating the XPS spectra of substituted ferrocene complexes and azobenzene isomers. For the ground electronic states of these molecules, the XPS spectra computed using the extended second-order MR-ADC method (MR-ADC(2)-X) are in a very good agreement with available experimental results. We further show that MR-ADC can be used as a tool for interpreting or predicting the results of time-resolved XPS measurements by simulating the core ionization spectra of azobenzene along its photoisomerization, including the XPS signatures of excited states and the minimum energy conical intersection. This work is the first in a series of publications reporting the efficient implementations of MR-ADC methods.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":null,"pages":null},"PeriodicalIF":2.7000,"publicationDate":"2024-07-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.4c03161","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
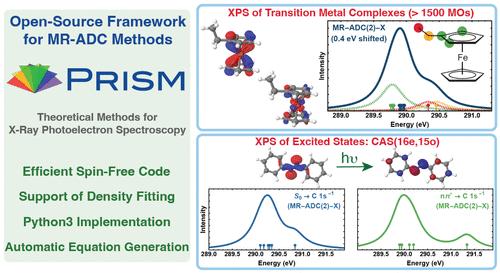
We present an efficient implementation of multireference algebraic diagrammatic construction theory (MR-ADC) for simulating core-ionized states and X-ray photoelectron spectra (XPS). Taking advantage of spin adaptation, automatic code generation, and density fitting, our implementation can perform calculations for molecules with more than 1500 molecular orbitals, incorporating static and dynamic correlation in the ground and excited electronic states. We demonstrate the capabilities of MR-ADC methods by simulating the XPS spectra of substituted ferrocene complexes and azobenzene isomers. For the ground electronic states of these molecules, the XPS spectra computed using the extended second-order MR-ADC method (MR-ADC(2)-X) are in a very good agreement with available experimental results. We further show that MR-ADC can be used as a tool for interpreting or predicting the results of time-resolved XPS measurements by simulating the core ionization spectra of azobenzene along its photoisomerization, including the XPS signatures of excited states and the minimum energy conical intersection. This work is the first in a series of publications reporting the efficient implementations of MR-ADC methods.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


