A K Larin, K M Klimina, V A Veselovsky, E I Olekhnovich, M D Morozov, D I Boldyreva, R A Yunes, A I Manolov, D E Fedorov, A V Pavlenko, Y S Galeeva, E V Starikova, E N Ilina
{"title":"An improved and extended dual-index multiplexed 16S rRNA sequencing for the Illumina HiSeq and MiSeq platform.","authors":"A K Larin, K M Klimina, V A Veselovsky, E I Olekhnovich, M D Morozov, D I Boldyreva, R A Yunes, A I Manolov, D E Fedorov, A V Pavlenko, Y S Galeeva, E V Starikova, E N Ilina","doi":"10.1186/s12863-024-01192-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Recent advancements in next-generation sequencing (NGS) technology have ushered in significant improvements in sequencing speed and data throughput, thereby enabling the simultaneous analysis of a greater number of samples within a single sequencing run. This technology has proven particularly valuable in the context of microbial community profiling, offering a powerful tool for characterizing the microbial composition at the species level within a given sample. This profiling process typically involves the sequencing of 16S ribosomal RNA (rRNA) gene fragments. By scaling up the analysis to accommodate a substantial number of samples, sometimes as many as 2,000, it becomes possible to achieve cost-efficiency and minimize the introduction of potential batch effects. Our study was designed with the primary objective of devising an approach capable of facilitating the comprehensive analysis of 1,711 samples sourced from diverse origins, including oropharyngeal swabs, mouth cavity swabs, dental swabs, and human fecal samples. This analysis was based on data obtained from 16S rRNA metagenomic sequencing conducted on the Illumina MiSeq and HiSeq sequencing platforms.</p><p><strong>Results: </strong>We have designed a custom set of 10-base pair indices specifically tailored for the preparation of libraries from amplicons derived from the V3-V4 region of the 16S rRNA gene. These indices are instrumental in the analysis of the microbial composition in clinical samples through sequencing on the Illumina MiSeq and HiSeq platforms. The utilization of our custom index set enables the consolidation of a significant number of libraries, enabling the efficient sequencing of these libraries in a single run.</p><p><strong>Conclusions: </strong>The unique array of 10-base pair indices that we have developed, in conjunction with our sequencing methodology, will prove highly valuable to laboratories engaged in sequencing on Illumina platforms or utilizing Illumina-compatible kits.</p>","PeriodicalId":72427,"journal":{"name":"BMC genomic data","volume":"25 1","pages":"8"},"PeriodicalIF":1.9000,"publicationDate":"2024-01-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10804484/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC genomic data","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12863-024-01192-3","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
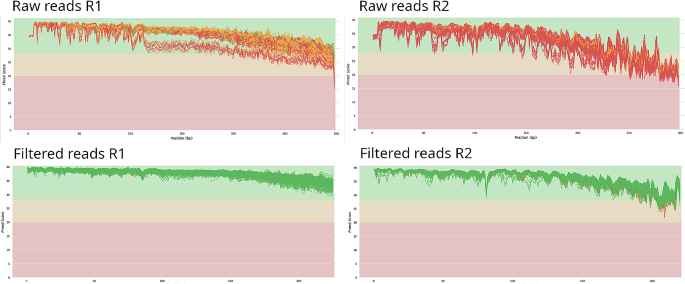
Background: Recent advancements in next-generation sequencing (NGS) technology have ushered in significant improvements in sequencing speed and data throughput, thereby enabling the simultaneous analysis of a greater number of samples within a single sequencing run. This technology has proven particularly valuable in the context of microbial community profiling, offering a powerful tool for characterizing the microbial composition at the species level within a given sample. This profiling process typically involves the sequencing of 16S ribosomal RNA (rRNA) gene fragments. By scaling up the analysis to accommodate a substantial number of samples, sometimes as many as 2,000, it becomes possible to achieve cost-efficiency and minimize the introduction of potential batch effects. Our study was designed with the primary objective of devising an approach capable of facilitating the comprehensive analysis of 1,711 samples sourced from diverse origins, including oropharyngeal swabs, mouth cavity swabs, dental swabs, and human fecal samples. This analysis was based on data obtained from 16S rRNA metagenomic sequencing conducted on the Illumina MiSeq and HiSeq sequencing platforms.
Results: We have designed a custom set of 10-base pair indices specifically tailored for the preparation of libraries from amplicons derived from the V3-V4 region of the 16S rRNA gene. These indices are instrumental in the analysis of the microbial composition in clinical samples through sequencing on the Illumina MiSeq and HiSeq platforms. The utilization of our custom index set enables the consolidation of a significant number of libraries, enabling the efficient sequencing of these libraries in a single run.
Conclusions: The unique array of 10-base pair indices that we have developed, in conjunction with our sequencing methodology, will prove highly valuable to laboratories engaged in sequencing on Illumina platforms or utilizing Illumina-compatible kits.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


