{"title":"Cluster-in-Molecule Local Correlation Method for Dispersion Interactions in Large Systems and Periodic Systems","authors":"Wei Li, Yuqi Wang, Zhigang Ni and Shuhua Li*, ","doi":"10.1021/acs.accounts.3c00538","DOIUrl":null,"url":null,"abstract":"<p >The noncovalent interactions, including dispersion interactions, control the structures and stabilities of complex chemical systems, including host–guest complexes and the adsorption process of molecules on the solid surfaces. The density functional theory (DFT) with empirical dispersion correction is now the working horse in many areas of applications. Post-Hartree–Fock (post-HF) methods have been well recognized to provide more accurate descriptions in a systematic way. However, traditional post-HF methods are mainly limited to small- or medium-sized systems, and their applications to periodic condensed phase systems are still very limited due to their expensive computational costs.</p><p >To extend post-HF calculations to large molecules, the cluster-in-molecule (CIM) local correlation approach has been established, allowing highly accurate electron correlation calculations that are routinely available for very large systems. In the CIM approach, the electron correlation energy of a large molecule could be obtained from electron correlation calculations on a series of clusters, each of which contains a subset of occupied and virtual localized molecular orbitals. The CIM method could be massively and efficiently parallelized on general computer clusters. The CIM method has been implemented at various electron correlation levels, including second-order Mo̷ller-Plesset perturbation theory (MP2), coupled cluster singles and doubles (CCSD), CCSD with perturbative triples correction [CCSD(T)], etc. The CIM-MP2 energy gradient algorithm was developed and applied to the geometry optimizations of large systems. The CIM method has also been extended to condensed-phase systems under periodic boundary conditions (PBC-CIM). For periodic systems, the correlation energy per unit cell could be evaluated with correlation energy contributions from a series of clusters that are built with localized Wannier functions.</p><p >CIM-based electron correlation calculations have been employed to investigate a number of chemical problems in which the dispersion interaction is important. CIM-based post-HF methods including CIM domain-based local pair natural orbital (DLPNO) CCSD(T) are applied to compute the relative or binding energies of biological systems or supramolecular complexes, the reaction barrier in a relatively complex chemical reaction. The CIM-MP2 method is used to obtain the optimized geometry of large systems. CIM-based post-HF calculations have also been used to compute the cohesive energies of molecular crystals and adsorption energies of molecules on the solid surfaces. The CIM and its PBC variant are expected to become a powerful theoretical tool for accurate calculations of the energies and structures for a broad range of large systems and condensed-phase systems with significant dispersion interactions.</p>","PeriodicalId":1,"journal":{"name":"Accounts of Chemical Research","volume":"56 23","pages":"3462–3474"},"PeriodicalIF":16.4000,"publicationDate":"2023-11-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Accounts of Chemical Research","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.accounts.3c00538","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
The noncovalent interactions, including dispersion interactions, control the structures and stabilities of complex chemical systems, including host–guest complexes and the adsorption process of molecules on the solid surfaces. The density functional theory (DFT) with empirical dispersion correction is now the working horse in many areas of applications. Post-Hartree–Fock (post-HF) methods have been well recognized to provide more accurate descriptions in a systematic way. However, traditional post-HF methods are mainly limited to small- or medium-sized systems, and their applications to periodic condensed phase systems are still very limited due to their expensive computational costs.
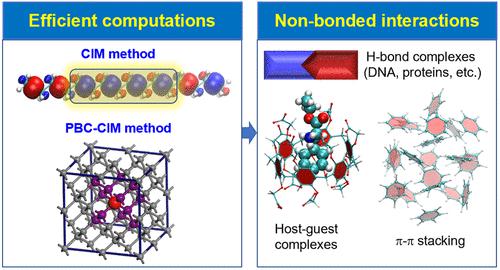
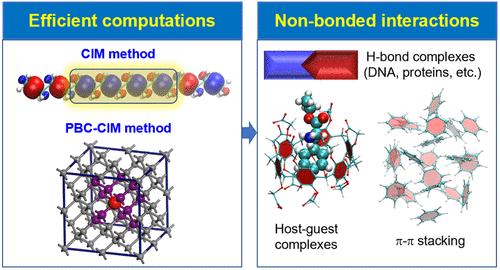
To extend post-HF calculations to large molecules, the cluster-in-molecule (CIM) local correlation approach has been established, allowing highly accurate electron correlation calculations that are routinely available for very large systems. In the CIM approach, the electron correlation energy of a large molecule could be obtained from electron correlation calculations on a series of clusters, each of which contains a subset of occupied and virtual localized molecular orbitals. The CIM method could be massively and efficiently parallelized on general computer clusters. The CIM method has been implemented at various electron correlation levels, including second-order Mo̷ller-Plesset perturbation theory (MP2), coupled cluster singles and doubles (CCSD), CCSD with perturbative triples correction [CCSD(T)], etc. The CIM-MP2 energy gradient algorithm was developed and applied to the geometry optimizations of large systems. The CIM method has also been extended to condensed-phase systems under periodic boundary conditions (PBC-CIM). For periodic systems, the correlation energy per unit cell could be evaluated with correlation energy contributions from a series of clusters that are built with localized Wannier functions.
CIM-based electron correlation calculations have been employed to investigate a number of chemical problems in which the dispersion interaction is important. CIM-based post-HF methods including CIM domain-based local pair natural orbital (DLPNO) CCSD(T) are applied to compute the relative or binding energies of biological systems or supramolecular complexes, the reaction barrier in a relatively complex chemical reaction. The CIM-MP2 method is used to obtain the optimized geometry of large systems. CIM-based post-HF calculations have also been used to compute the cohesive energies of molecular crystals and adsorption energies of molecules on the solid surfaces. The CIM and its PBC variant are expected to become a powerful theoretical tool for accurate calculations of the energies and structures for a broad range of large systems and condensed-phase systems with significant dispersion interactions.
期刊介绍:
Accounts of Chemical Research presents short, concise and critical articles offering easy-to-read overviews of basic research and applications in all areas of chemistry and biochemistry. These short reviews focus on research from the author’s own laboratory and are designed to teach the reader about a research project. In addition, Accounts of Chemical Research publishes commentaries that give an informed opinion on a current research problem. Special Issues online are devoted to a single topic of unusual activity and significance.
Accounts of Chemical Research replaces the traditional article abstract with an article "Conspectus." These entries synopsize the research affording the reader a closer look at the content and significance of an article. Through this provision of a more detailed description of the article contents, the Conspectus enhances the article's discoverability by search engines and the exposure for the research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


