Ruimin Wang, Binli Wang, Abubakar Sadiq Abdullahi, Hongjun Fan
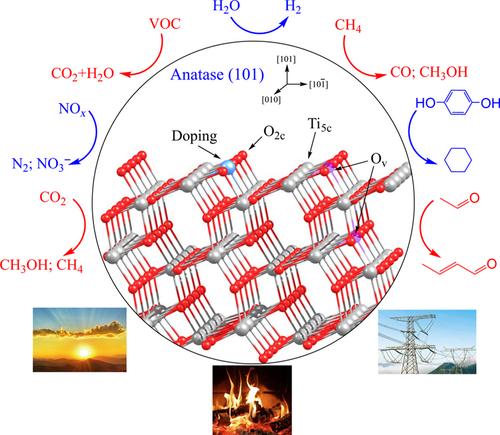
{"title":"Understanding the prototype catalyst TiO2 surface with the help of density functional theory calculation","authors":"Ruimin Wang, Binli Wang, Abubakar Sadiq Abdullahi, Hongjun Fan","doi":"10.1002/wcms.1686","DOIUrl":null,"url":null,"abstract":"<p>Titanium dioxide (TiO<sub>2</sub>) is one of the most technologically promising oxides with a broad range of catalytic and photocatalytic activities. Theoretical modeling, especially density functional theory calculations, has been extensively carried out to understand the geometric structure, electronic structure, reactivity, and reaction mechanisms of TiO<sub>2</sub> systems, as well as to develop new catalysts with improved performances. This review summarizes the recent theoretical progress on the well-defined surfaces of TiO<sub>2</sub> crystalline, and focuses on the structures, adsorptions, and reactions on the surface and at the interface. The theoretical methods and models, surface defects, surface doping, water splitting and H<sub>2</sub> evolution, methanol conversion, CO<sub>2</sub> reduction and CO oxidation, SO<sub><i>x</i></sub> and NO<sub><i>x</i></sub> degradation, CH<sub>4</sub> conversion, organic pollutant degradation, C<span></span>H bond activation and C<span></span>C bond formation, dye sensitization, as well as the applications of TiO<sub>2</sub> in some other fields, have been discussed in detail.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 1","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1686","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
Titanium dioxide (TiO2) is one of the most technologically promising oxides with a broad range of catalytic and photocatalytic activities. Theoretical modeling, especially density functional theory calculations, has been extensively carried out to understand the geometric structure, electronic structure, reactivity, and reaction mechanisms of TiO2 systems, as well as to develop new catalysts with improved performances. This review summarizes the recent theoretical progress on the well-defined surfaces of TiO2 crystalline, and focuses on the structures, adsorptions, and reactions on the surface and at the interface. The theoretical methods and models, surface defects, surface doping, water splitting and H2 evolution, methanol conversion, CO2 reduction and CO oxidation, SOx and NOx degradation, CH4 conversion, organic pollutant degradation, CH bond activation and CC bond formation, dye sensitization, as well as the applications of TiO2 in some other fields, have been discussed in detail.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


